Male fertility
MartinGarlovsky
2022-07-27
Last updated: 2025-04-02
Checks: 7 0
Knit directory: mito_age_fert/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20230213) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 5ebfaed. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.DS_Store
Ignored: data/.DS_Store
Ignored: output/.DS_Store
Untracked files:
Untracked: README.html
Untracked: check_lines.R
Untracked: code/data_wrangling.R
Untracked: code/female_predictions_13-Feb-2025.sh
Untracked: code/female_rate_predictions.R
Untracked: code/plotting_Script.R
Untracked: data/Data_raw_emmely.csv
Untracked: data/SNP_data/
Untracked: data/defence.csv
Untracked: data/male_fertility.csv
Untracked: data/mito_34sigdiffSNPs_consensus_incl_colnames.csv
Untracked: data/mito_mt_copy_number.xlsx
Untracked: data/mito_mt_seq_major_alleles_sig_snptable.csv
Untracked: data/mito_mt_seq_sig_annotated.csv
Untracked: data/mito_mt_seq_sig_annotated.vcf
Untracked: data/offence.csv
Untracked: data/rawdata_PCA.csv
Untracked: data/snp-gene.txt
Untracked: data/sperm_metabolic_rate.csv
Untracked: data/sperm_viability.csv
Untracked: data/wrangled/
Untracked: figures/
Untracked: output/SNP_clusters.csv
Untracked: output/anova_tables/
Untracked: output/bod_brm.rds
Untracked: output/female_rate_dredge.rds
Untracked: output/female_rates_bb.Rdata
Untracked: output/female_rates_boot.Rdata
Untracked: output/female_rates_poly.Rdata
Untracked: output/fst_tabs.csv
Untracked: output/male_fec_dredge.rds
Untracked: output/male_hatch_dredge.rds
Untracked: output/male_hatch_dredge_reduced.rds
Untracked: output/sperm_met_dredge.rds
Untracked: output/viab_ctrl_dredge.rds
Untracked: output/viab_trt_dredge.rds
Untracked: snp_matrix_dobler.csv
Unstaged changes:
Modified: analysis/SNP_clusters.Rmd
Deleted: analysis/sperm_comp.Rmd
Modified: data/README.md
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/male_fertility.Rmd) and
HTML (docs/male_fertility.html) files. If you’ve configured
a remote Git repository (see ?wflow_git_remote), click on
the hyperlinks in the table below to view the files as they were in that
past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 5ebfaed | MartinGarlovsky | 2025-04-02 | wflow_publish(c("analysis/body_size.Rmd", "analysis/female_fertility.Rmd", |
| html | 0ffce9c | MartinGarlovsky | 2024-10-10 | Build site. |
| Rmd | dfaa300 | MartinGarlovsky | 2024-10-10 | wflow_publish("analysis/male_fertility.Rmd") |
| html | a7bae31 | MartinGarlovsky | 2024-10-09 | Build site. |
| Rmd | 5f3a2a7 | MartinGarlovsky | 2024-10-09 | wflow_publish("analysis/male_fertility.Rmd") |
Load packages
library(tidyverse)
library(lme4)
library(DHARMa)
library(emmeans)
library(kableExtra)
library(knitrhooks) # install with devtools::install_github("nathaneastwood/knitrhooks")
library(showtext)
library(conflicted)
select <- dplyr::select
filter <- dplyr::filter
output_max_height() # a knitrhook option
options(stringsAsFactors = FALSE)
# colour palettes
met.pal <- MetBrewer::met.brewer('Johnson')
met3 <- met.pal[c(1, 3, 5)]
# set contrasts
options(contrasts = c("contr.sum", "contr.poly"))Load data
# fecundity and hatching success
male_fert_filt <- read.csv('data/wrangled/male_fertility.csv') %>%
mutate(age = fct_relevel(age, c("young","old")),
mito_snp = as.factor(mito_snp),
coevolved = if_else(mito == nuclear, "matched", "mismatched"))
#xtabs(~ mito + nuclear, data = male_fert)
# sperm viability
sperm_vib_filt <- read.csv("data/wrangled/sperm_viability_wrangled.csv") %>%
mutate(age = fct_relevel(age, c("young","old")),
mito_snp = as.factor(mito_snp),
coevolved = if_else(mito == nuclear, "matched", "mismatched"))
#xtabs(~ mito + nuclear, data = sperm_vib_filt)
# sperm metabolism
sperm_met <- read.csv("data/wrangled/sperm_met_wrangled.csv") %>%
mutate(age = fct_relevel(age, c("young","old")),
mito_snp = as.factor(mito_snp),
p.a2 = a2/100, # convert to percentage proportion
coevolved = if_else(mito == nuclear, "matched", "mismatched"))
#xtabs(~ mito + nuclear, data = sperm_met)
# sperm competition data
## P1
defence <- read.csv("data/wrangled/sperm_defence.csv", header = TRUE) %>%
mutate(mito_snp = as.factor(mito_snp),
Day = ordered(Day),
coevolved = if_else(mito == nuclear, "matched", "mismatched"))
## P2
offence <- read.csv("data/wrangled/sperm_offence.csv", header = TRUE) %>%
mutate(mito_snp = as.factor(mito_snp),
Day = ordered(Day),
coevolved = if_else(mito == nuclear, "matched", "mismatched"))> Fecundity
male_fert_filt %>%
group_by(mito, nuclear, age) %>%
summarise(mn = mean(total_eggs),
se = sd(total_eggs)/sqrt(n()),
s95 = se * 1.96) %>%
ggplot(aes(x = mito, y = mn, fill = nuclear)) +
geom_point(data = male_fert_filt,
aes(y = total_eggs, colour = nuclear), alpha = .25,
position = position_jitterdodge(dodge.width = .5, jitter.width = .1)) +
geom_errorbar(aes(ymin = mn - s95, ymax = mn + s95),
width = .25, position = position_dodge(width = .5)) +
geom_point(size = 3, pch = 21, position = position_dodge(width = .5)) +
labs(y = 'Partner fecundity') +
facet_wrap(~ age, scales = 'free_x') +
scale_colour_manual(values = met3) +
scale_fill_manual(values = met3) +
theme_bw() +
theme() +
NULL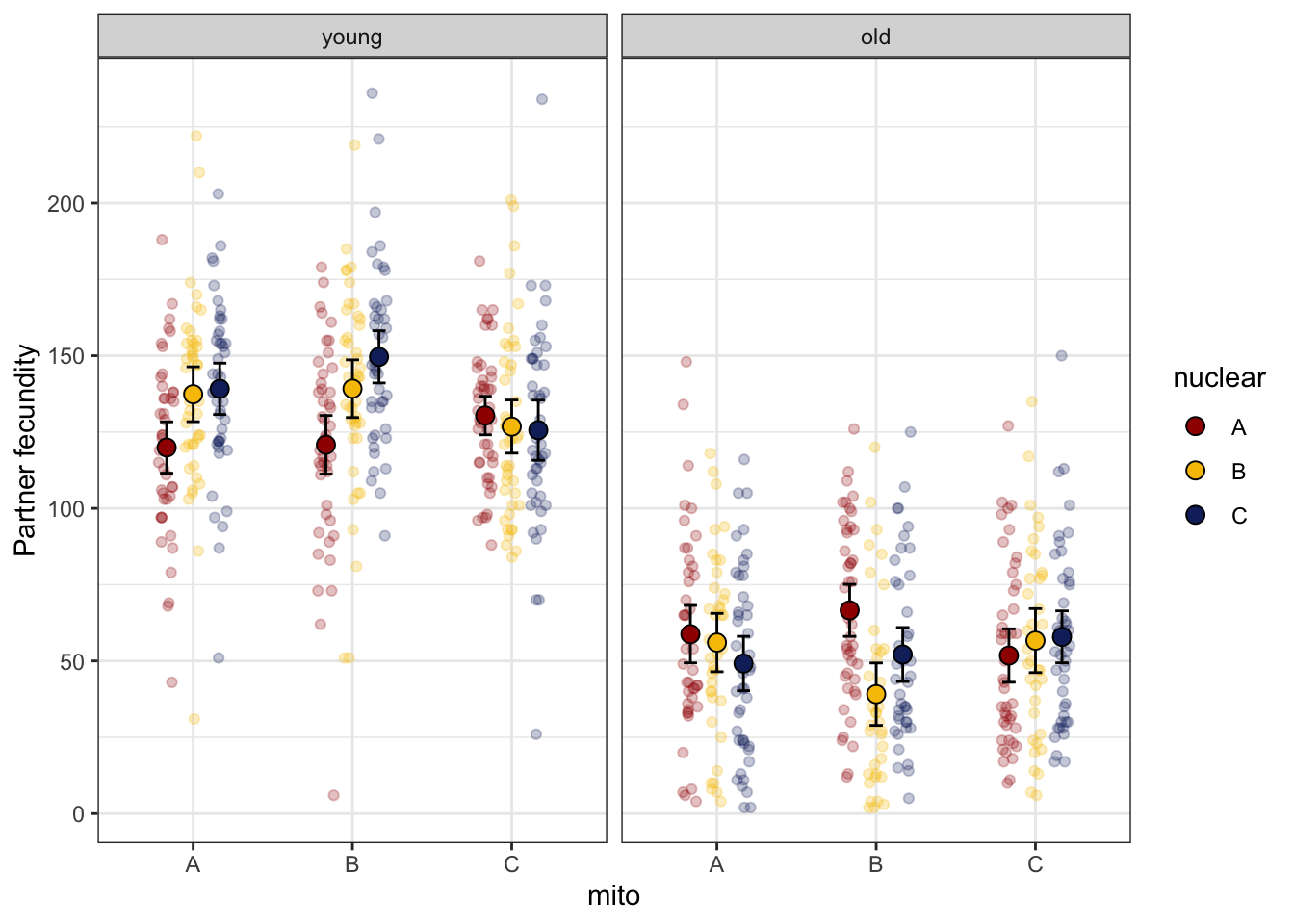
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
hist(male_fert_filt$total_eggs, breaks = 50)
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
# fit linear model - model diagnostics look better?
male_fec_test <- lmerTest::lmer(total_eggs ~ mito * nuclear * age + (1|LINE:age),
data = male_fert_filt, REML = TRUE)
anova(male_fec_test, type = "III", ddf = "Kenward-Roger") %>%
broom::tidy() %>%
as_tibble() %>% #write_csv("output/anova_tables/male_fecundity.csv") %>% # save anova table for supp. tables
kable(digits = 3,
caption = 'Type III Analysis of Variance Table with Kenward-Roger`s method') %>%
kable_styling(full_width = FALSE)| term | sumsq | meansq | NumDF | DenDF | statistic | p.value |
|---|---|---|---|---|---|---|
| mito | 655.057 | 327.529 | 2 | 36.119 | 0.375 | 0.690 |
| nuclear | 1328.203 | 664.101 | 2 | 36.089 | 0.761 | 0.474 |
| age | 628683.021 | 628683.021 | 1 | 36.120 | 720.654 | 0.000 |
| mito:nuclear | 3177.393 | 794.348 | 4 | 36.089 | 0.911 | 0.468 |
| mito:age | 2413.910 | 1206.955 | 2 | 36.119 | 1.384 | 0.264 |
| nuclear:age | 9125.522 | 4562.761 | 2 | 36.089 | 5.230 | 0.010 |
| mito:nuclear:age | 12241.283 | 3060.321 | 4 | 36.089 | 3.508 | 0.016 |
#summary(male_fec_test)
fec_emm <- emmeans(male_fec_test, ~ mito * nuclear * age)
#pairs(fec_emm, simple = list("age", "mito", "nuclear"))
bind_rows(pairs(fec_emm, simple = list("age", "mito", "nuclear"))$`simple contrasts for age` %>% broom::tidy(),
pairs(fec_emm, simple = list("age", "mito", "nuclear"))$`simple contrasts for mito` %>% broom::tidy() %>%
rename(p.value = adj.p.value),
pairs(fec_emm, simple = list("age", "mito", "nuclear"))$`simple contrasts for nuclear` %>% broom::tidy() %>%
rename(p.value = adj.p.value)) %>%
mutate(p.val = ifelse(p.value < 0.001, '< 0.001', round(p.value, 3))) %>%
select(-p.value, -term) %>% relocate(age, .before = contrast) %>%
kable(digits = 3,
caption = 'Posthoc Tukey tests to compare which groups differ') %>%
kable_styling(full_width = FALSE) %>%
kableExtra::group_rows("Age", 1, 9) %>%
kableExtra::group_rows("Mito contrasts", 10, 27) %>%
kableExtra::group_rows("Nuclear contrasts", 28, 45)| mito | nuclear | age | contrast | null.value | estimate | std.error | df | statistic | p.val |
|---|---|---|---|---|---|---|---|---|---|
| Age | |||||||||
| A | A | NA | young - old | 0 | 61.156 | 8.602 | 34.548 | 7.109 | < 0.001 |
| B | A | NA | young - old | 0 | 54.244 | 8.602 | 34.548 | 6.306 | < 0.001 |
| C | A | NA | young - old | 0 | 78.724 | 8.628 | 34.960 | 9.124 | < 0.001 |
| A | B | NA | young - old | 0 | 81.208 | 8.825 | 37.887 | 9.202 | < 0.001 |
| B | B | NA | young - old | 0 | 100.156 | 8.882 | 39.205 | 11.276 | < 0.001 |
| C | B | NA | young - old | 0 | 70.090 | 8.888 | 39.184 | 7.886 | < 0.001 |
| A | C | NA | young - old | 0 | 90.000 | 8.602 | 34.548 | 10.463 | < 0.001 |
| B | C | NA | young - old | 0 | 97.573 | 8.684 | 35.849 | 11.236 | < 0.001 |
| C | C | NA | young - old | 0 | 67.711 | 8.602 | 34.548 | 7.871 | < 0.001 |
| Mito contrasts | |||||||||
| NA | A | young | A - B | 0 | -0.889 | 8.602 | 34.548 | -0.103 | 0.994 |
| NA | A | young | A - C | 0 | -10.467 | 8.602 | 34.548 | -1.217 | 0.452 |
| NA | A | young | B - C | 0 | -9.578 | 8.602 | 34.548 | -1.113 | 0.512 |
| NA | B | young | A - B | 0 | -1.822 | 8.602 | 34.548 | -0.212 | 0.976 |
| NA | B | young | A - C | 0 | 10.600 | 8.602 | 34.548 | 1.232 | 0.443 |
| NA | B | young | B - C | 0 | 12.422 | 8.602 | 34.548 | 1.444 | 0.33 |
| NA | C | young | A - B | 0 | -10.467 | 8.602 | 34.548 | -1.217 | 0.452 |
| NA | C | young | A - C | 0 | 13.556 | 8.602 | 34.548 | 1.576 | 0.269 |
| NA | C | young | B - C | 0 | 24.022 | 8.602 | 34.548 | 2.793 | 0.022 |
| NA | A | old | A - B | 0 | -7.800 | 8.602 | 34.548 | -0.907 | 0.64 |
| NA | A | old | A - C | 0 | 7.102 | 8.628 | 34.960 | 0.823 | 0.691 |
| NA | A | old | B - C | 0 | 14.902 | 8.628 | 34.960 | 1.727 | 0.21 |
| NA | B | old | A - B | 0 | 17.126 | 9.098 | 42.719 | 1.882 | 0.156 |
| NA | B | old | A - C | 0 | -0.517 | 9.104 | 42.692 | -0.057 | 0.998 |
| NA | B | old | B - C | 0 | -17.643 | 9.160 | 44.100 | -1.926 | 0.143 |
| NA | C | old | A - B | 0 | -2.894 | 8.684 | 35.849 | -0.333 | 0.941 |
| NA | C | old | A - C | 0 | -8.733 | 8.602 | 34.548 | -1.015 | 0.572 |
| NA | C | old | B - C | 0 | -5.839 | 8.684 | 35.849 | -0.672 | 0.781 |
| Nuclear contrasts | |||||||||
| A | NA | young | A - B | 0 | -17.444 | 8.602 | 34.548 | -2.028 | 0.121 |
| A | NA | young | A - C | 0 | -19.222 | 8.602 | 34.548 | -2.235 | 0.079 |
| A | NA | young | B - C | 0 | -1.778 | 8.602 | 34.548 | -0.207 | 0.977 |
| B | NA | young | A - B | 0 | -18.378 | 8.602 | 34.548 | -2.136 | 0.097 |
| B | NA | young | A - C | 0 | -28.800 | 8.602 | 34.548 | -3.348 | 0.005 |
| B | NA | young | B - C | 0 | -10.422 | 8.602 | 34.548 | -1.212 | 0.454 |
| C | NA | young | A - B | 0 | 3.622 | 8.602 | 34.548 | 0.421 | 0.907 |
| C | NA | young | A - C | 0 | 4.800 | 8.602 | 34.548 | 0.558 | 0.843 |
| C | NA | young | B - C | 0 | 1.178 | 8.602 | 34.548 | 0.137 | 0.99 |
| A | NA | old | A - B | 0 | 2.608 | 8.825 | 37.887 | 0.295 | 0.953 |
| A | NA | old | A - C | 0 | 9.622 | 8.602 | 34.548 | 1.119 | 0.509 |
| A | NA | old | B - C | 0 | 7.015 | 8.825 | 37.887 | 0.795 | 0.708 |
| B | NA | old | A - B | 0 | 27.533 | 8.882 | 39.205 | 3.100 | 0.01 |
| B | NA | old | A - C | 0 | 14.528 | 8.684 | 35.849 | 1.673 | 0.229 |
| B | NA | old | B - C | 0 | -13.005 | 8.962 | 40.582 | -1.451 | 0.325 |
| C | NA | old | A - B | 0 | -5.012 | 8.913 | 39.619 | -0.562 | 0.841 |
| C | NA | old | A - C | 0 | -6.213 | 8.628 | 34.960 | -0.720 | 0.753 |
| C | NA | old | B - C | 0 | -1.201 | 8.888 | 39.184 | -0.135 | 0.99 |
>>> Results
>>>> Reaction norms
# reaction norms
male_fecundity_norm <- emmeans(male_fec_test, ~ mito * nuclear * age) %>% as_tibble() %>%
ggplot(aes(x = nuclear, y = emmean, fill = mito)) +
geom_line(aes(group = mito, colour = mito), position = position_dodge(width = .5)) +
# geom_errorbar(aes(ymin = lower.CL, ymax = upper.CL),
# width = .25, position = position_dodge(width = .5)) +
geom_point(size = 3, pch = 21, position = position_dodge(width = .5)) +
labs(y = 'Partner fecundity (EMM ± 95% CI)') +
scale_colour_manual(values = met3) +
scale_fill_manual(values = met3) +
facet_wrap(~ age) +
theme_bw() +
theme() +
NULL
male_fecundity_norm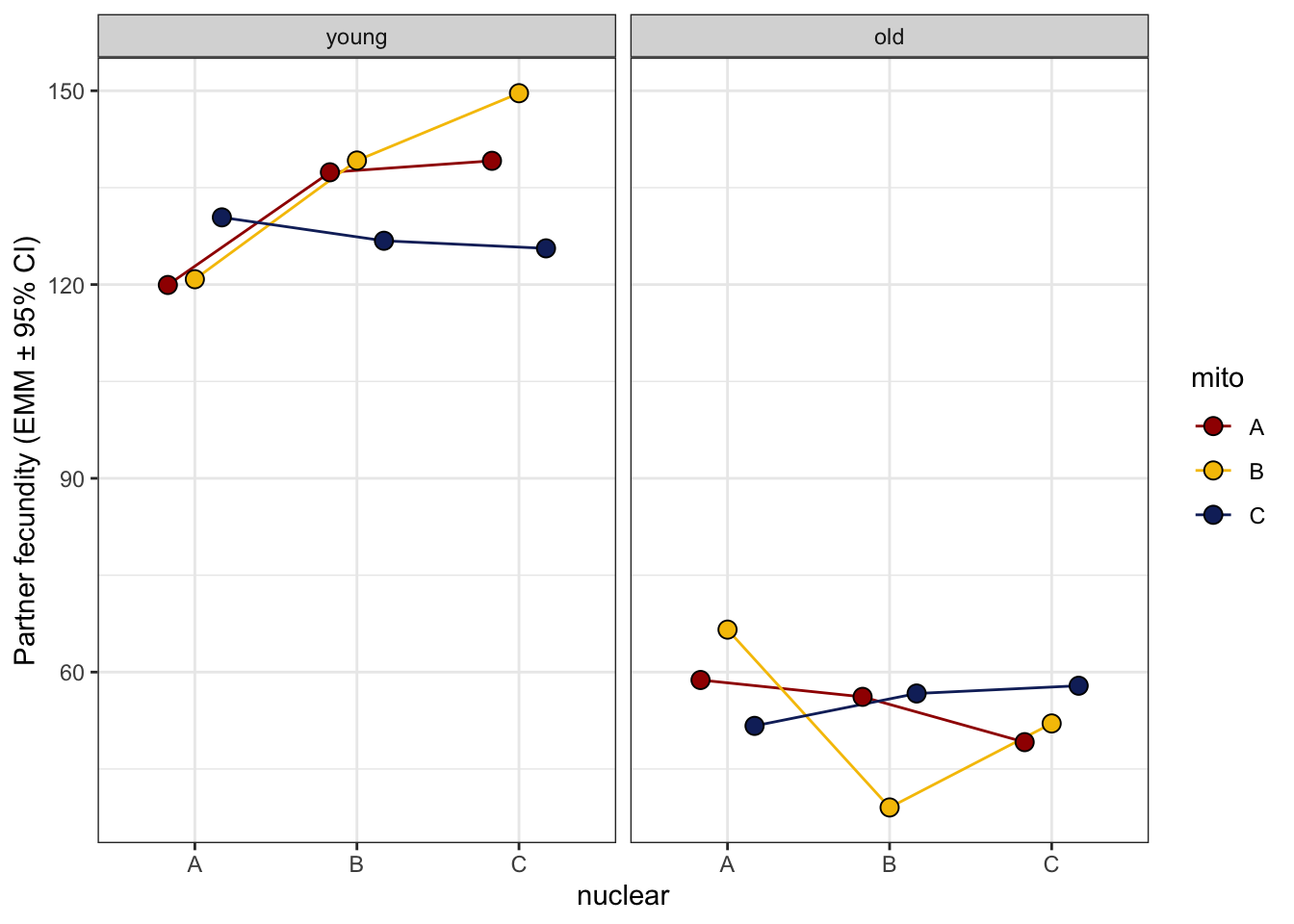
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
>>>> Raw data
male_fecundity_raw <- emmeans(male_fec_test, ~ mito * nuclear * age) %>% as_tibble() %>%
ggplot(aes(x = nuclear, y = emmean, fill = mito)) +
geom_jitter(data = male_fert_filt,
aes(y = total_eggs, colour = mito),
position = position_jitterdodge(dodge.width = .5, jitter.width = .1),
size = 0.75, alpha = .1) +
geom_jitter(data = male_fert_filt %>%
group_by(LINE, age) %>%
summarise(mn = mean(total_eggs)) %>%
separate(LINE, into = c("mito", "nuclear", NA), sep = "(?<=.)", remove = FALSE),
aes(y = mn, colour = mito),
position = position_jitterdodge(dodge.width = .5, jitter.width = .1),
alpha = .85, size = 2) +
geom_errorbar(aes(ymin = lower.CL, ymax = upper.CL),
width = .25, position = position_dodge(width = .5)) +
geom_point(size = 3, pch = 21, position = position_dodge(width = .5)) +
labs(y = 'Partner fecundity (EMM ± 95% CIs)') +
scale_colour_manual(values = met3) +
scale_fill_manual(values = met3) +
facet_wrap(~ age) +
theme_bw() +
theme() +
geom_text(data = male_fert_filt %>% group_by(mito, nuclear, age) %>% count(), aes(y = -5, label = n),
size = 2, position = position_dodge(width = .5)) +
#ggsave('figures/male_fecundity_raw.pdf', height = 4, width = 8, dpi = 600, useDingbats = FALSE) +
NULL
male_fecundity_raw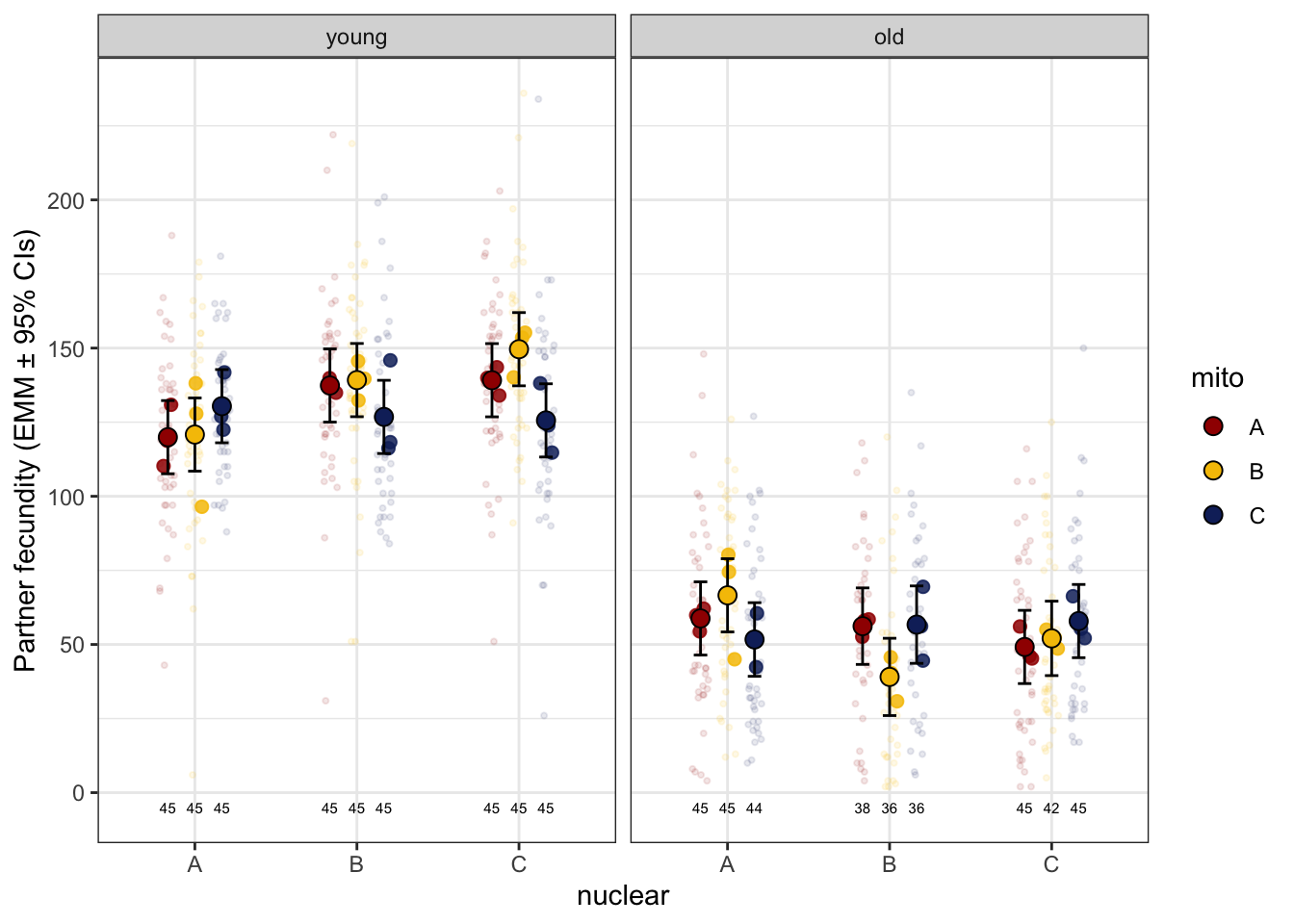
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
>>> Matched vs. mismatched
male_fec_coevo <- lmerTest::lmer(total_eggs ~ coevolved * age + (1 + age|LINE),
data = male_fert_filt, REML = TRUE)
anova(male_fec_coevo, type = "III", ddf = "Kenward-Roger") %>%
broom::tidy() %>%
as_tibble()# A tibble: 3 × 7 term sumsq meansq NumDF DenDF statistic p.value1 coevolved 1256. 1256. 1 24.9 1.44 2.42e- 1 2 age 442492. 442492. 1 24.9 507. 4.32e-18 3 coevolved:age 151. 151. 1 24.9 0.173 6.81e- 1
#summary(male_fec_coevo)>>> Mito-type analysis
# fit linear model
male_fec1_snp <- lmerTest::lmer(total_eggs ~ mito_snp * nuclear * age + (1|LINE:age),
data = male_fert_filt, REML = TRUE)
anova(male_fec1_snp, type = "III", ddf = "Kenward-Roger") %>% broom::tidy() %>%
as_tibble() %>%
kable(digits = 3,
caption = 'Type III Analysis of Variance Table with Kenward-Roger`s method') %>%
kable_styling(full_width = FALSE)| term | sumsq | meansq | NumDF | DenDF | statistic | p.value |
|---|---|---|---|---|---|---|
| mito_snp | 1680.257 | 210.032 | 8 | 17.998 | 0.241 | 0.977 |
| nuclear | 169.779 | 84.889 | 2 | 18.050 | 0.097 | 0.908 |
| age | 254992.935 | 254992.935 | 1 | 17.837 | 292.215 | 0.000 |
| mito_snp:nuclear | 4216.698 | 602.385 | 7 | 18.144 | 0.690 | 0.679 |
| mito_snp:age | 3795.329 | 474.416 | 8 | 17.998 | 0.544 | 0.809 |
| nuclear:age | 5043.513 | 2521.757 | 2 | 18.050 | 2.890 | 0.082 |
| mito_snp:nuclear:age | 4183.266 | 597.609 | 7 | 18.144 | 0.685 | 0.683 |
#car::Anova(male_fec1_snp, type = "III")
fec_emm_snp <- emmeans(male_fec1_snp, ~ mito_snp * nuclear * age)
male_fecundity_snp <- fec_emm_snp %>% as_tibble() %>% drop_na() %>%
ggplot(aes(x = nuclear, y = emmean, fill = mito_snp)) +
geom_jitter(data = male_fert_filt,
aes(y = total_eggs, colour = mito_snp),
position = position_jitterdodge(dodge.width = .5, jitter.width = .1),
alpha = .25) +
geom_errorbar(aes(ymin = lower.CL, ymax = upper.CL),
width = .25, position = position_dodge(width = .5)) +
geom_point(size = 3, pch = 21, position = position_dodge(width = .5)) +
labs(y = 'Partner fecundity (EMM ± 95% CIs)') +
facet_wrap(~ age) +
scale_colour_viridis_d(option = "H") +
scale_fill_viridis_d(option = "H") +
theme_bw() +
theme() +
#ggsave('figures/male_fecundity_snp.pdf', height = 4, width = 8.5, dpi = 600, useDingbats = FALSE) +
NULL
male_fecundity_snp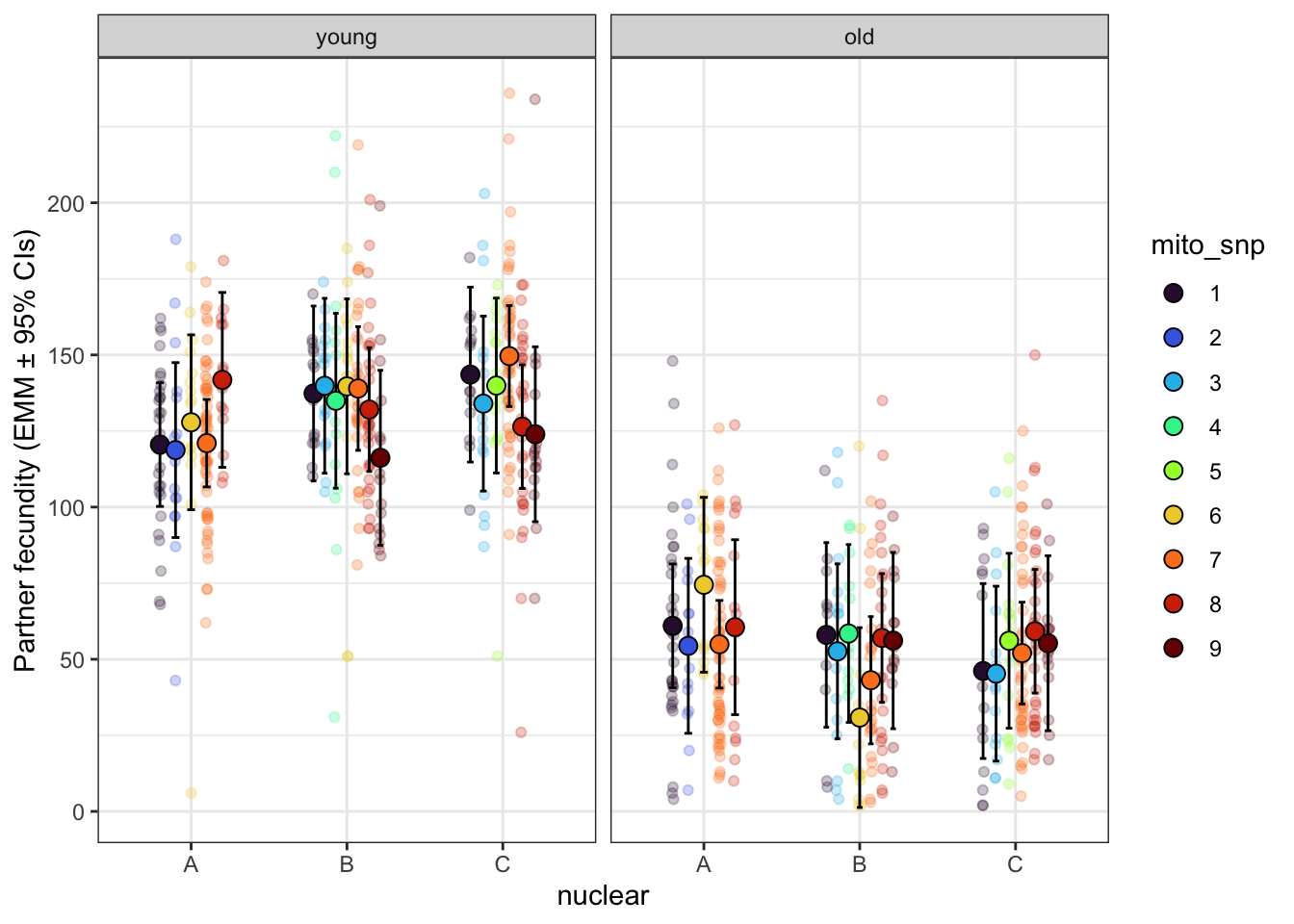
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
> Hatching success
Hatching success, our proxy for fertilisation success, shows different patterns in young and old males. In old males there is a very bimodal distribtuion, i.e., some males are fertile while others are completely sterile. Therefore, we model young and old males separately.
male_fert_filt %>%
group_by(mito, nuclear, age) %>%
summarise(mn = mean(fertilised),
se = sd(fertilised)/sqrt(n()),
s95 = se * 1.96) %>%
ggplot(aes(x = nuclear, y = mn, fill = mito)) +
geom_point(data = male_fert_filt,
aes(y = fertilised, colour = mito), alpha = .25,
position = position_jitterdodge(dodge.width = .5, jitter.width = .1)) +
geom_errorbar(aes(ymin = mn - s95, ymax = mn + s95),
width = .25, position = position_dodge(width = .5)) +
geom_point(size = 3, pch = 21, position = position_dodge(width = .5)) +
labs(y = 'Hatching success') +
coord_cartesian(ylim = c(0, 1)) +
facet_wrap(~ age, scales = 'free_x') +
scale_colour_manual(values = met3) +
scale_fill_manual(values = met3) +
theme_bw() +
theme() +
#ggsave('figures/male_hatchingsuccess_all.pdf', height = 4, width = 9, dpi = 600, useDingbats = FALSE) +
NULL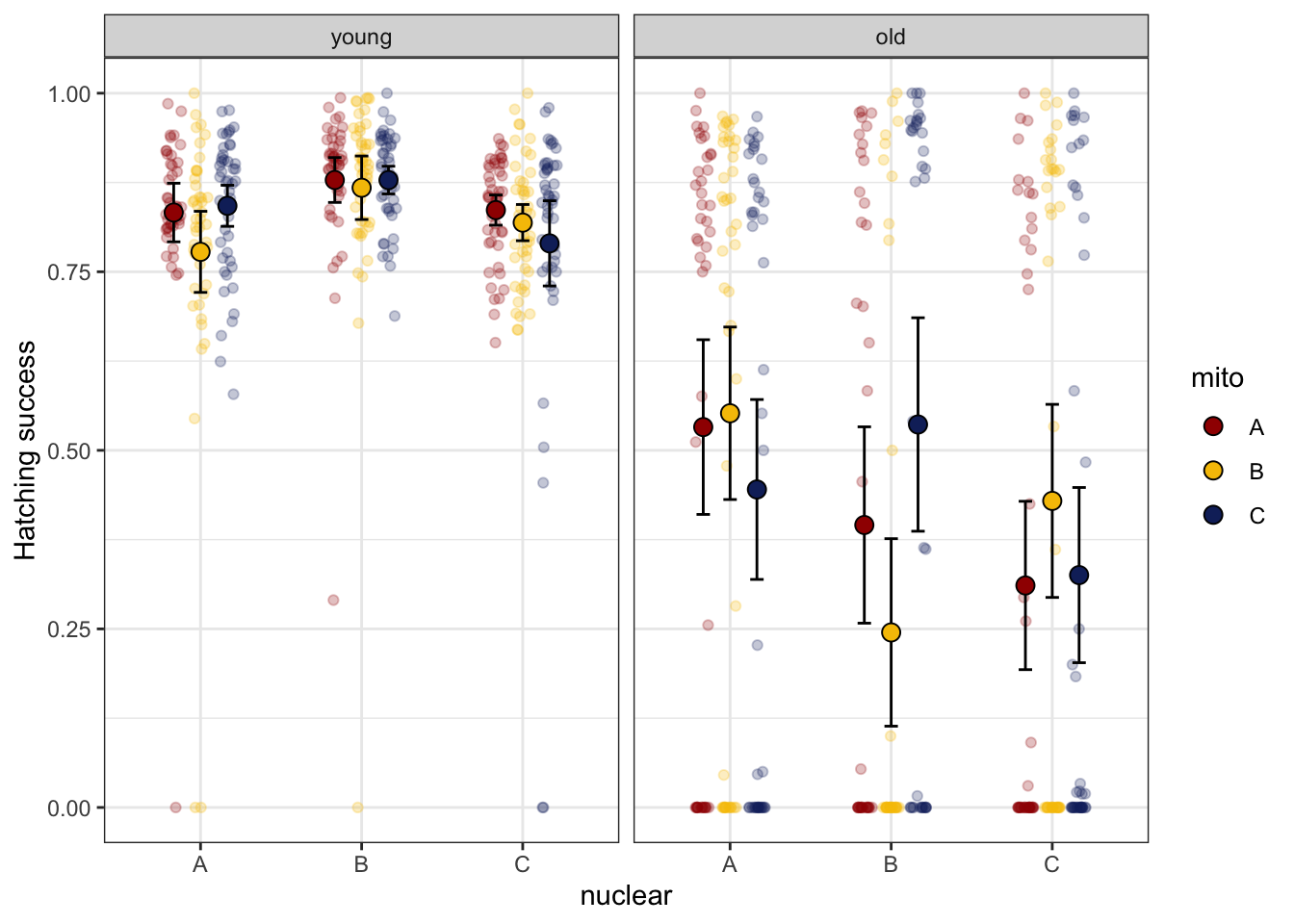
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
#xtabs(~ mtn + age, data = male_fert_filt)>>> Young males
In young males we model the proportion of eggs that hatch.
##model young only
hatch2 <- glmer(cbind(total_offs, total_fail) ~ mito * nuclear + (1|LINE) + (1|OLRE),
data = male_fert_filt %>% filter(age == "young"),
family = 'binomial')>>>> Check model diagnostics
performance::check_model(hatch2)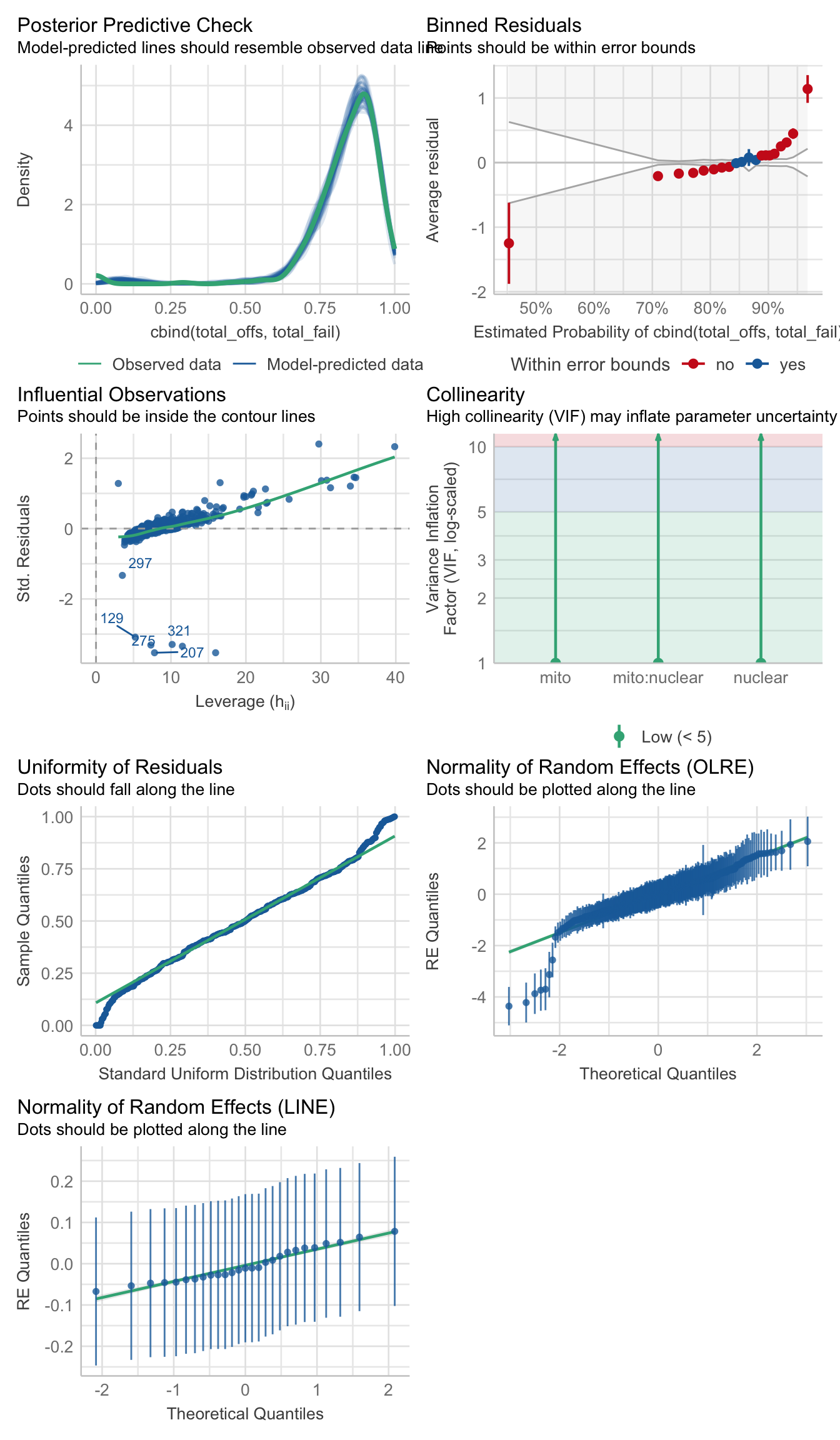
performance::check_overdispersion(hatch2)# Overdispersion test
dispersion ratio = 1.020
p-value = 0.688
testDispersion(hatch2)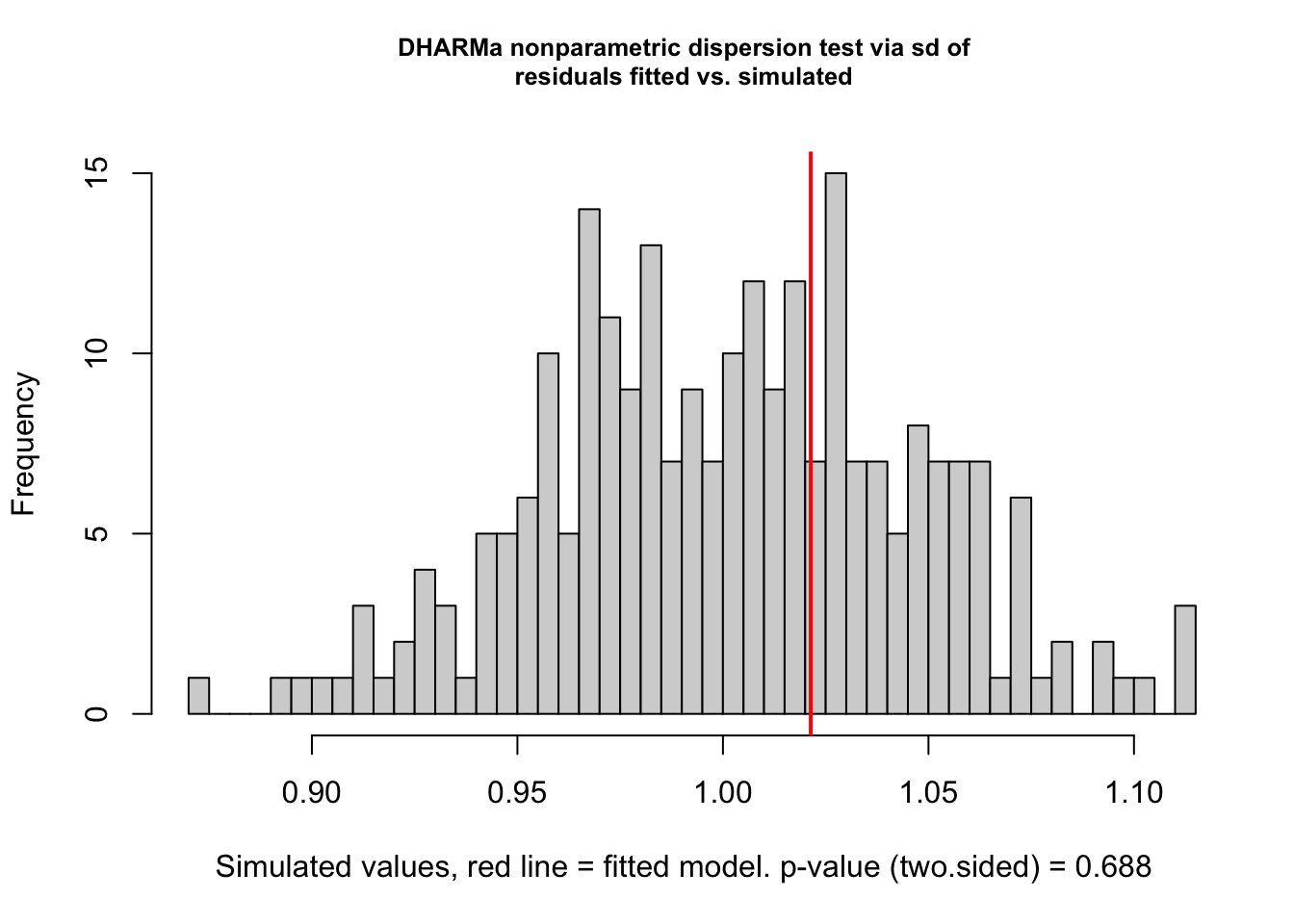
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
DHARMa nonparametric dispersion test via sd of residuals fitted vs.
simulated
data: simulationOutput
dispersion = 1.02, p-value = 0.688
alternative hypothesis: two.sided
simulationOutput <- simulateResiduals(fittedModel = hatch2, plot = FALSE)
hist(residuals(simulationOutput))
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
hist(residuals(simulationOutput, quantileFunction = qnorm, outlierValues = c(-7,7)), breaks = 50)
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
plot(simulationOutput)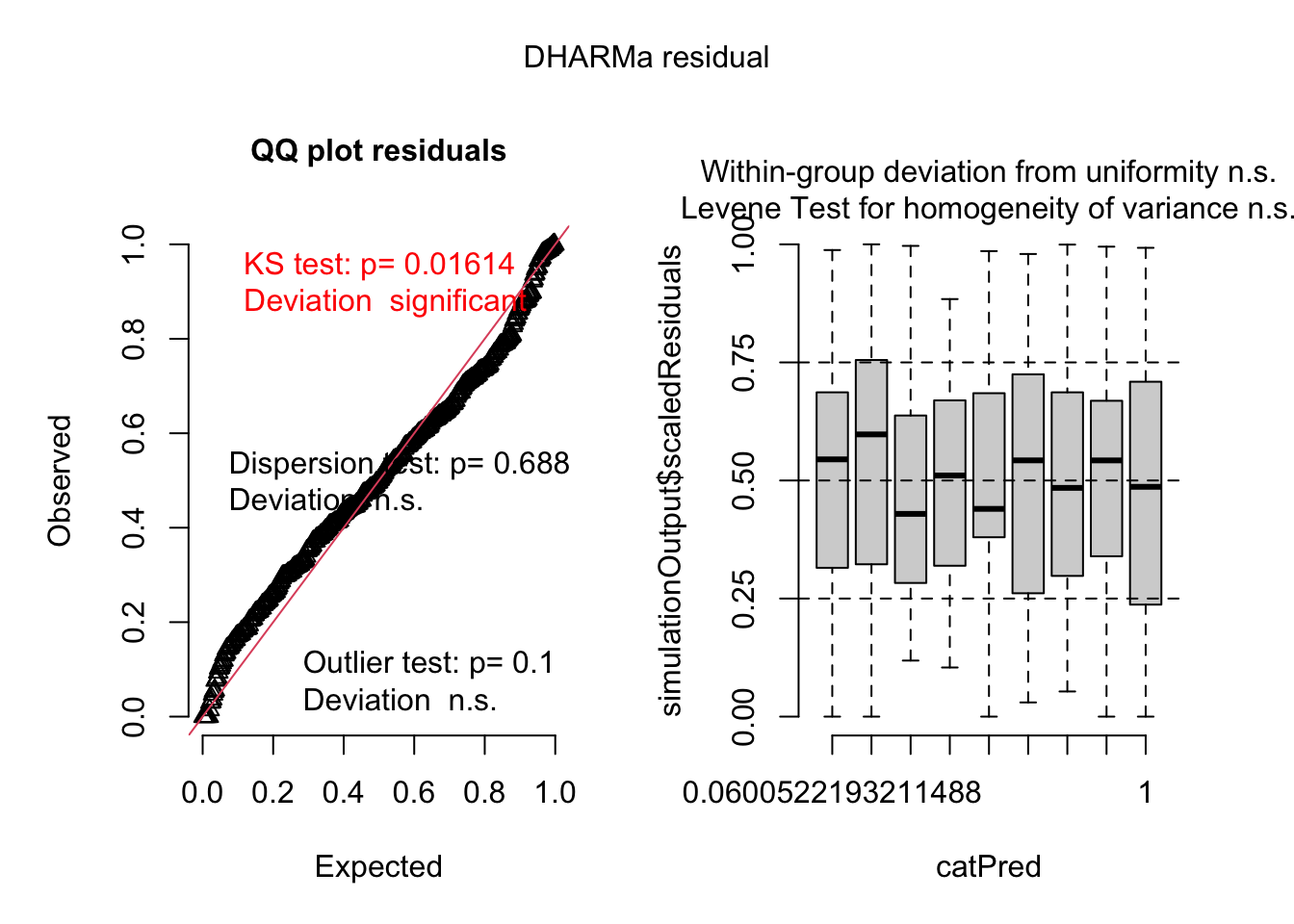
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
>>> Results
car::Anova(hatch2, type = "3") %>%
broom::tidy() %>%
as_tibble() %>% #write_csv("output/anova_tables/male_fert_young.csv") %>% # save anova table for supp. tables
kable(digits = 3,
caption = 'Analysis of Deviance Table (Type III Wald chisquare tests)') %>%
kable_styling(full_width = FALSE)| term | statistic | df | p.value |
|---|---|---|---|
| (Intercept) | 1422.684 | 1 | 0.000 |
| mito | 1.540 | 2 | 0.463 |
| nuclear | 26.190 | 2 | 0.000 |
| mito:nuclear | 5.198 | 4 | 0.268 |
#summary(hatch2)
hatch_emm2 <- emmeans(hatch2, ~ mito * nuclear)
emmeans(hatch2, pairwise ~ nuclear, adjust = "tukey")$contrasts %>% as_tibble() %>%
kable(digits = 3,
caption = 'Posthoc Tukey tests for comparison between nuclear genotypes') %>%
kable_styling(full_width = FALSE)| contrast | estimate | SE | df | z.ratio | p.value |
|---|---|---|---|---|---|
| A - B | -0.514 | 0.119 | Inf | -4.310 | 0.000 |
| A - C | 0.028 | 0.118 | Inf | 0.239 | 0.969 |
| B - C | 0.542 | 0.119 | Inf | 4.563 | 0.000 |
>>>> Reaction norms
# reaction norms
hatchingsuccess_young_norm <- emmeans(hatch2, ~ mito * nuclear, type = 'response') %>% as_tibble() %>%
ggplot(aes(x = nuclear, y = prob, fill = mito)) +
geom_line(aes(group = mito, colour = mito), position = position_dodge(width = .5)) +
geom_point(size = 3, pch = 21, position = position_dodge(width = .5)) +
labs(y = '% Hatching success (EMM ± 95% CI)') +
scale_colour_manual(values = met3) +
scale_fill_manual(values = met3) +
theme_bw() +
theme() +
NULL
hatchingsuccess_young_norm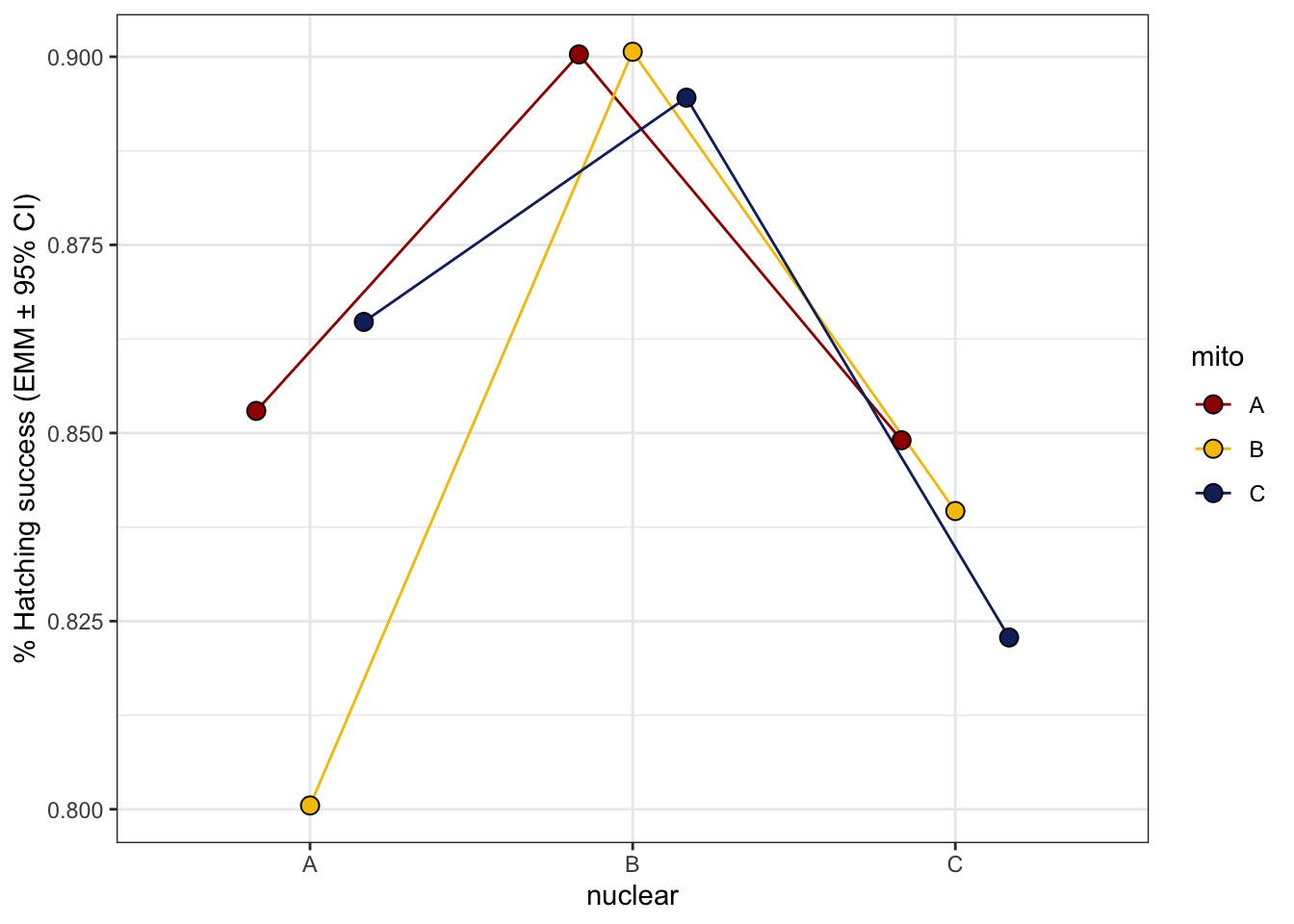
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
>>>> Raw data with means
hatchingsuccess_young_raw <- emmeans(hatch2, ~ mito * nuclear, type = 'response') %>% as_tibble() %>%
ggplot(aes(x = nuclear, y = prob, fill = mito)) +
geom_jitter(data = male_fert_filt %>% filter(age == "young"),
aes(y = fertilised, colour = mito),
position = position_jitterdodge(dodge.width = .5, jitter.width = .1),
alpha = .25) +
geom_jitter(data = male_fert_filt %>% filter(age == "young") %>%
group_by(LINE, age) %>%
summarise(mn = mean(fertilised)) %>%
separate(LINE, into = c("mito", "nuclear", NA), sep = "(?<=.)", remove = FALSE),
aes(y = mn, colour = mito),
position = position_jitterdodge(dodge.width = .5, jitter.width = .1),
alpha = .85, size = 2) +
geom_errorbar(aes(ymin = asymp.LCL, ymax = asymp.UCL),
width = .25, position = position_dodge(width = .5)) +
geom_point(size = 3, pch = 21, position = position_dodge(width = .5)) +
labs(y = '% Hatching success (EMM ± 95% CIs)') +
scale_colour_manual(values = met3) +
scale_fill_manual(values = met3) +
theme_bw() +
theme() +
geom_text(data = male_fert_filt %>% filter(age == "young") %>%
group_by(mito, nuclear) %>% count(), aes(y = -0.04, label = n),
size = 2, position = position_dodge(width = .5)) +
#ggsave('figures/hatchingsuccess_young_raw.pdf', height = 4, width = 5, dpi = 600, useDingbats = FALSE) +
NULL
hatchingsuccess_young_raw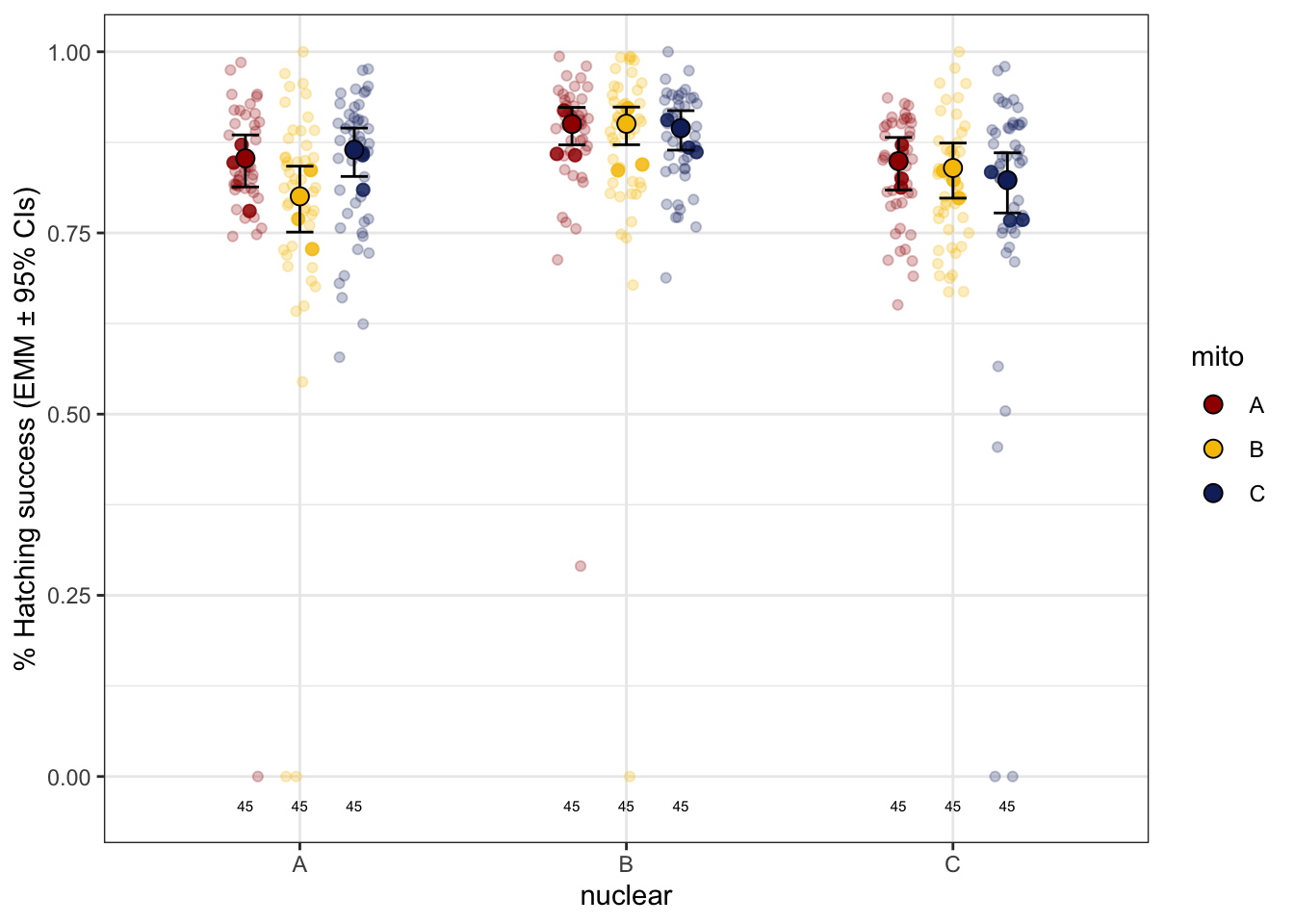
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
>>> Matched vs. mismatched
hatch_young_coevo <- glmer(cbind(total_offs, total_fail) ~ coevolved + (1|LINE) + (1|OLRE),
data = male_fert_filt %>% filter(age == "young"),
family = 'binomial')
performance::check_model(hatch_young_coevo)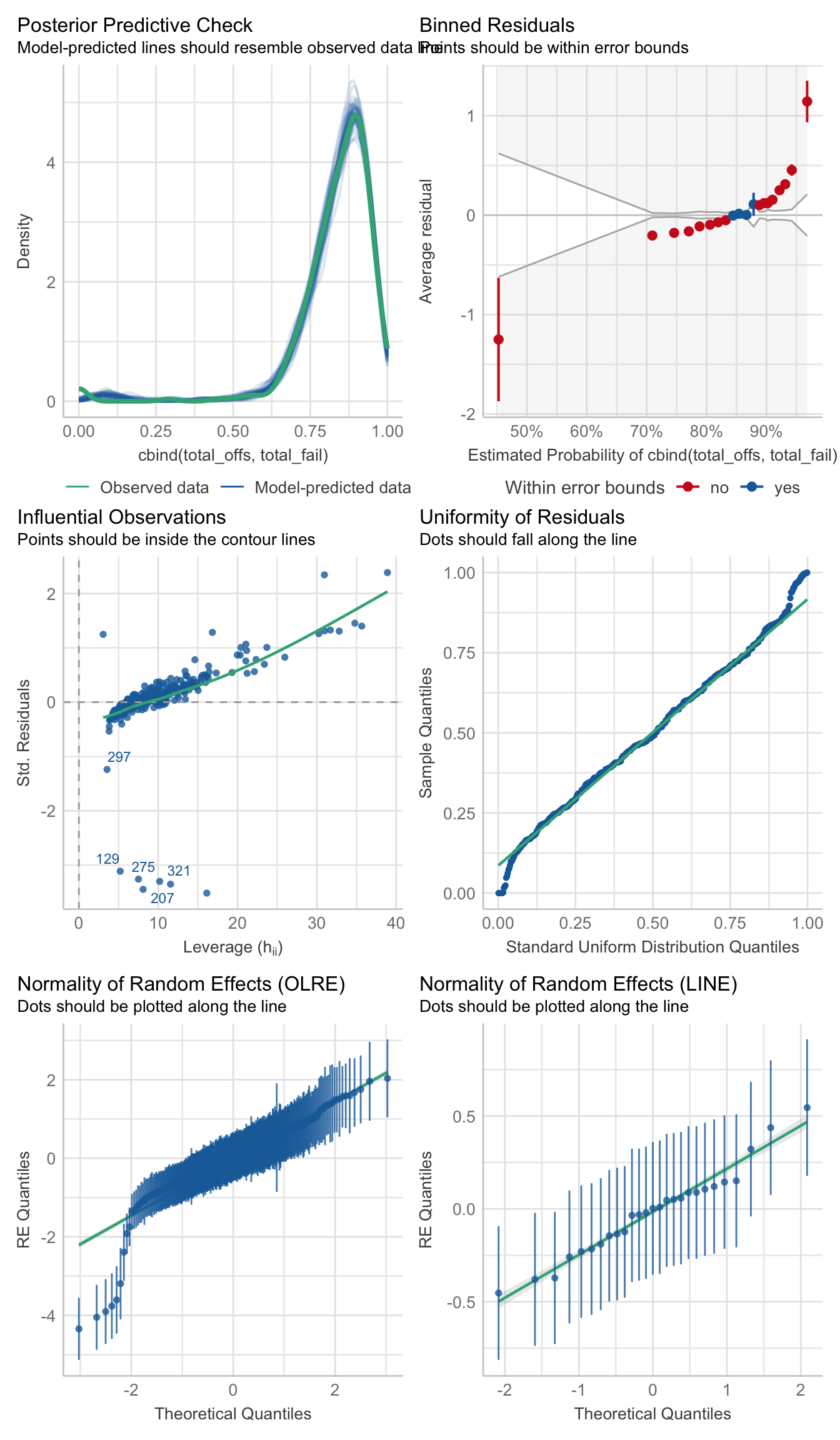
car::Anova(hatch_young_coevo, type = "3") %>% broom::tidy() %>%
as_tibble() %>%
kable(digits = 3,
caption = 'Analysis of Deviance Table (Type III Wald chisquare tests)') %>%
kable_styling(full_width = FALSE)| term | statistic | df | p.value |
|---|---|---|---|
| (Intercept) | 565.895 | 1 | 0.000 |
| coevolved | 0.002 | 1 | 0.964 |
>>> Mito-type analysis
young_fert_snp <- glmer(cbind(total_offs, total_fail) ~ mito_snp * nuclear + (1|LINE) + (1|OLRE),
data = male_fert_filt %>% filter(age == "young"),
family = "binomial",
control = glmerControl(optimizer = "bobyqa", optCtrl = list(maxfun = 50000)))
performance::check_model(young_fert_snp)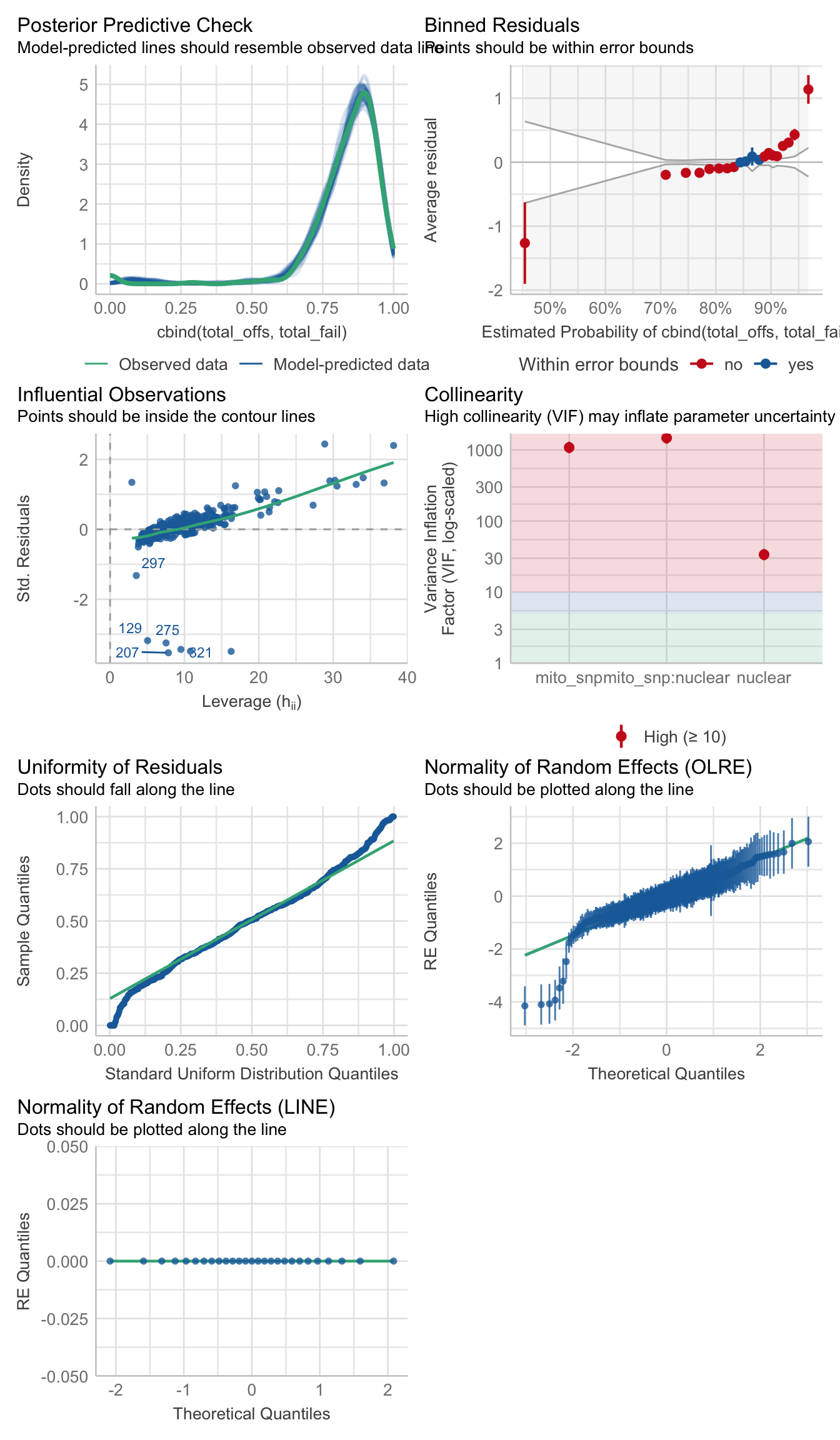
car::Anova(young_fert_snp, type = "3") %>% broom::tidy() %>%
as_tibble() %>%
kable(digits = 3,
caption = 'Analysis of Deviance Table (Type III Wald chisquare tests)') %>%
kable_styling(full_width = FALSE)| term | statistic | df | p.value |
|---|---|---|---|
| (Intercept) | 118.245 | 1 | 0.000 |
| mito_snp | 10.256 | 8 | 0.248 |
| nuclear | 2.496 | 2 | 0.287 |
| mito_snp:nuclear | 9.013 | 7 | 0.252 |
>>> Old males
In old males we will model fertility as a binary variable - whether a male was fertile (1) or sterile (0). A stacked barchart of the binary response again clearly shows most young males are fertile, but a significant proporiton of old males fail to fertilise any eggs.
# add a new variable giving a binary fertile (1) or sterile (0) based on whether the proportion fertilised was 0 or greater than 0.
male_sterilty <- male_fert_filt %>%
mutate(sterility = if_else(fertilised == 0, 'sterile', 'fertile'),
sterilbin = if_else(fertilised == 0, 0, 1),
OLRE = 1:nrow(.))
male_sterilty %>%
group_by(mito, nuclear, age, sterility) %>%
count() %>%
ggplot(aes(x = nuclear, y = n, fill = sterility)) +
geom_col(position = 'fill') +
labs(y = "Proportion of males") +
facet_grid(mito ~ age)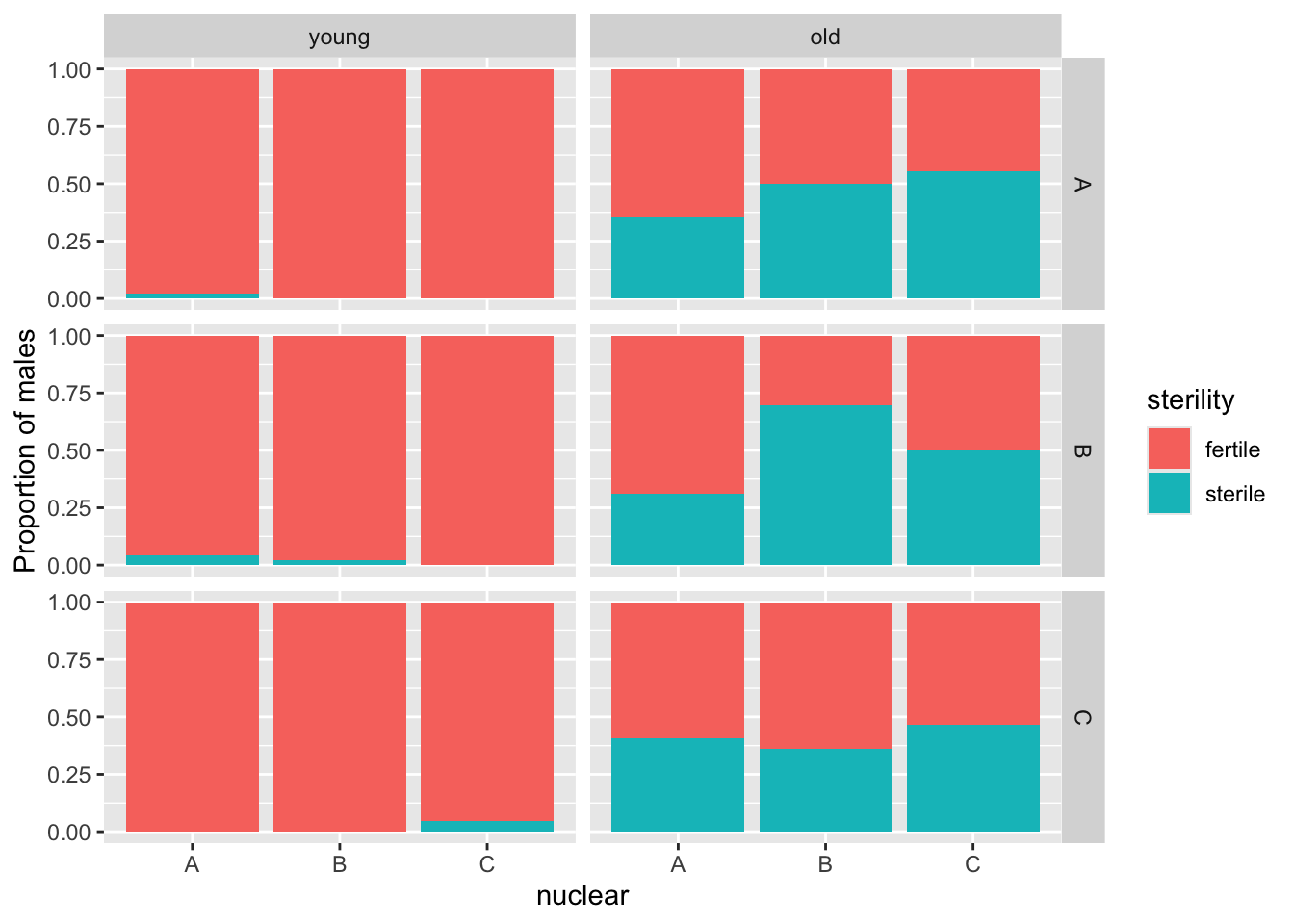
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
# model old males only
ster_md <- glmer(sterilbin ~ mito * nuclear + (1|LINE), family = 'binomial',
data = male_sterilty %>% filter(age == "old"))
#summary(ster_md)>>>> Check diagnostics
performance::check_model(ster_md)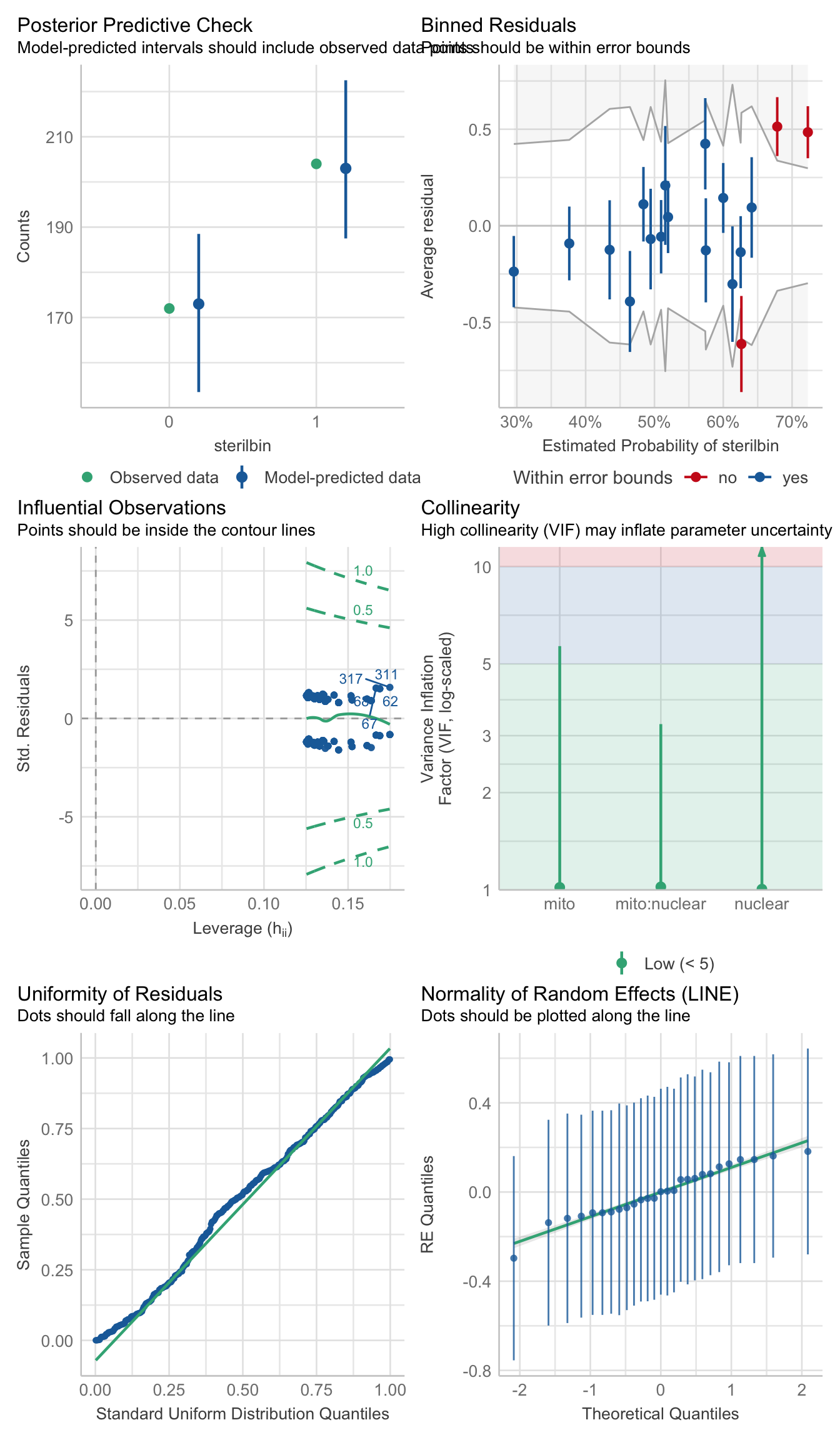
performance::check_overdispersion(ster_md)# Overdispersion test
dispersion ratio = 1.005
p-value = 0.848
>>>> Results
car::Anova(ster_md, type = "3") %>%
broom::tidy() %>%
as_tibble() %>% #write_csv("output/anova_tables/male_fert_old.csv") %>% # save anova table for supp. tables
kable(digits = 3,
caption = 'Analysis of Deviance Table (Type III Wald chisquare tests)') %>%
kable_styling(full_width = FALSE)| term | statistic | df | p.value |
|---|---|---|---|
| (Intercept) | 1.819 | 1 | 0.177 |
| mito | 1.640 | 2 | 0.440 |
| nuclear | 6.819 | 2 | 0.033 |
| mito:nuclear | 6.935 | 4 | 0.139 |
ster_emm <- emmeans(ster_md, ~ mito * nuclear)
emmeans(ster_emm, pairwise ~ nuclear, adjust = "tukey")$contrasts %>% as_tibble() %>%
kable(digits = 3,
caption = 'Posthoc Tukey tests for comparison between nuclear genotypes') %>%
kable_styling(full_width = FALSE)| contrast | estimate | SE | df | z.ratio | p.value |
|---|---|---|---|---|---|
| A - B | 0.680 | 0.298 | Inf | 2.280 | 0.059 |
| A - C | 0.629 | 0.282 | Inf | 2.226 | 0.067 |
| B - C | -0.051 | 0.294 | Inf | -0.175 | 0.983 |
>>>>> Reaction norms
# reaction norms
hatchingsuccess_old_norm <- emmeans(ster_emm, ~ mito * nuclear, type = 'response') %>% as_tibble() %>%
ggplot(aes(x = nuclear, y = prob, fill = mito)) +
geom_line(aes(group = mito, colour = mito), position = position_dodge(width = .5)) +
geom_point(size = 3, pch = 21, position = position_dodge(width = .5)) +
labs(y = 'Proportion fertile (EMM ± 95% CI)') +
scale_colour_manual(values = met3) +
scale_fill_manual(values = met3) +
theme_bw() +
theme() +
NULL
hatchingsuccess_old_norm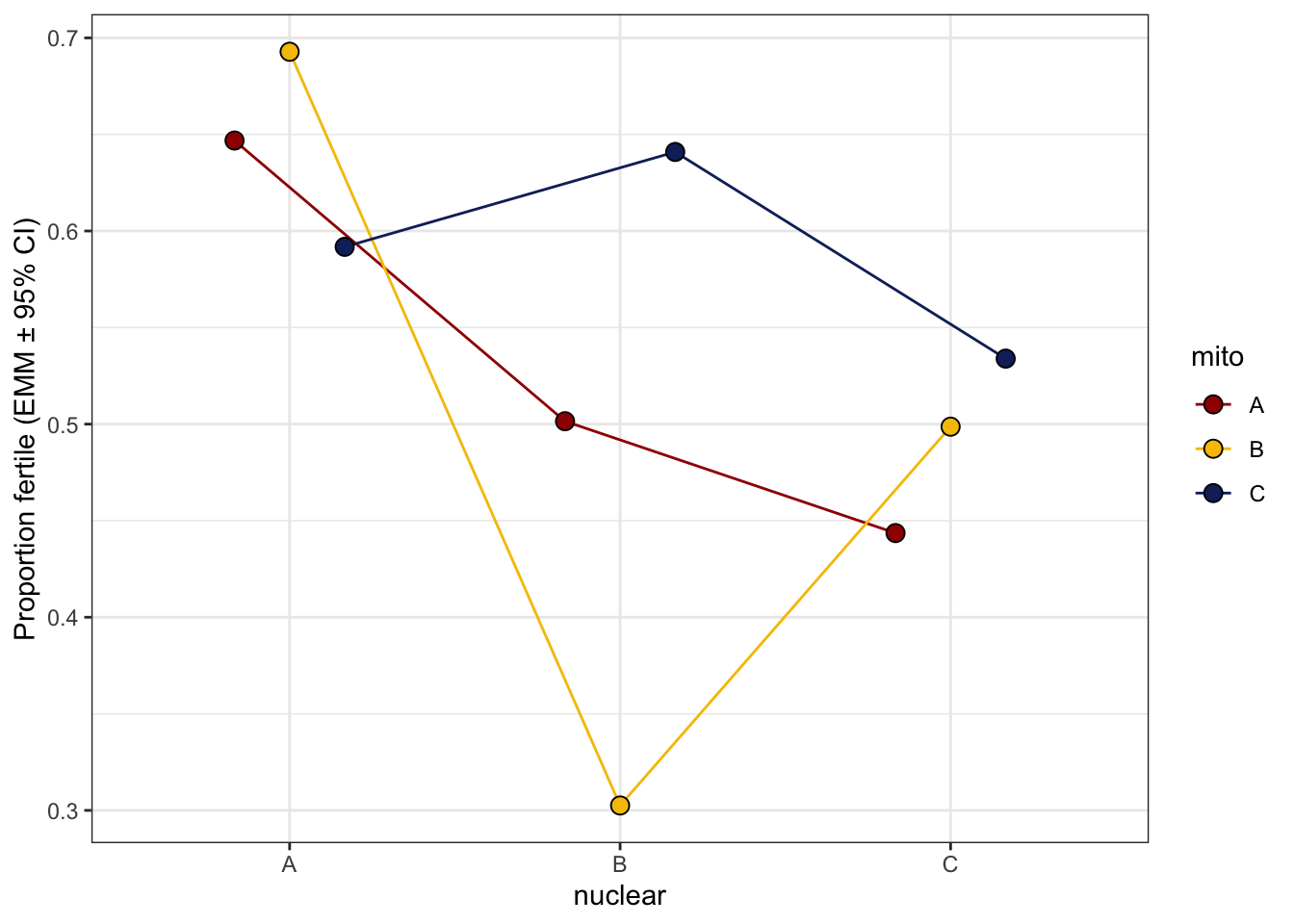
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
>>>>> Raw data and means
hatchingsuccess_old_raw <- emmeans(ster_emm, ~ mito * nuclear, type = 'response') %>% as_tibble() %>%
ggplot(aes(x = nuclear, y = prob, fill = mito)) +
geom_jitter(data = male_fert_filt %>% filter(age == "old"),
aes(y = fertilised, colour = mito),
position = position_jitterdodge(dodge.width = .5, jitter.width = .1),
alpha = .25) +
geom_jitter(data = male_fert_filt %>% filter(age == "old") %>%
group_by(LINE, age) %>%
summarise(mn = mean(fertilised)) %>%
separate(LINE, into = c("mito", "nuclear", NA), sep = "(?<=.)", remove = FALSE),
aes(y = mn, colour = mito),
position = position_jitterdodge(dodge.width = .5, jitter.width = .1),
alpha = .85, size = 2) +
geom_errorbar(aes(ymin = asymp.LCL, ymax = asymp.UCL),
width = .25, position = position_dodge(width = .5)) +
geom_point(size = 3, pch = 21, position = position_dodge(width = .5)) +
labs(y = 'Proportion fertile (EMM ± 95% CIs)') +
scale_colour_manual(values = met3) +
scale_fill_manual(values = met3) +
theme_bw() +
theme() +
geom_text(data = male_fert_filt %>% filter(age == "young") %>%
group_by(mito, nuclear) %>% count(), aes(y = -0.04, label = n),
size = 2, position = position_dodge(width = .5)) +
#ggsave('figures/hatchingsuccess_old_raw.pdf', height = 4, width = 5, dpi = 600, useDingbats = FALSE) +
NULL
hatchingsuccess_old_raw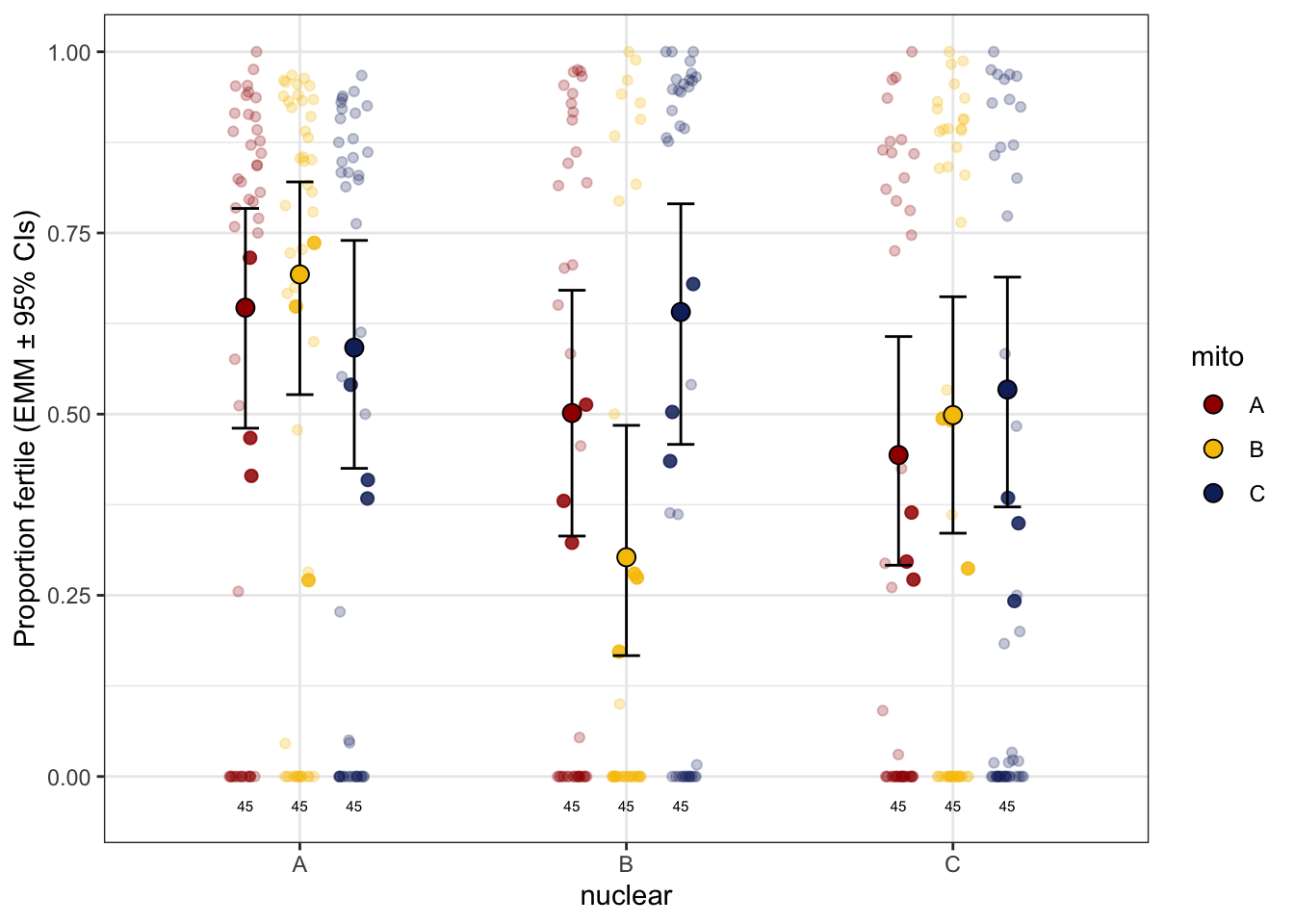
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
>>> Matched vs. mismatched
ster_coevo <- glmer(sterilbin ~ coevolved + (1|LINE), family = 'binomial',
data = male_sterilty %>% filter(age == "old"))
car::Anova(ster_coevo, type = "3") %>% broom::tidy() %>%
as_tibble() %>%
kable(digits = 3,
caption = 'Analysis of Deviance Table (Type III Wald chisquare tests)') %>%
kable_styling(full_width = FALSE)| term | statistic | df | p.value |
|---|---|---|---|
| (Intercept) | 0.749 | 1 | 0.387 |
| coevolved | 0.649 | 1 | 0.420 |
>>> Mito-type analysis
ster_md_snp <- glmer(sterilbin ~ mito_snp * nuclear + (1|LINE),
family = 'binomial',
data = male_sterilty %>% filter(age == "old"))
#summary(ster_md)
car::Anova(ster_md_snp, type = "3") %>% broom::tidy() %>%
as_tibble() %>%
kable(digits = 3,
caption = 'Analysis of Deviance Table (Type III Wald chisquare tests)') %>%
kable_styling(full_width = FALSE)| term | statistic | df | p.value |
|---|---|---|---|
| (Intercept) | 1.084 | 1 | 0.298 |
| mito_snp | 5.792 | 8 | 0.671 |
| nuclear | 3.084 | 2 | 0.214 |
| mito_snp:nuclear | 9.564 | 7 | 0.215 |
>>>>> combined plot
comb_fert <- bind_rows(
emmeans(hatch2, ~ mito * nuclear, type = 'response') %>% as_tibble() %>% mutate(age = "Young"),
emmeans(ster_emm, ~ mito * nuclear, type = 'response') %>% as_tibble() %>% mutate(age = "Old")) %>%
mutate(age = fct_relevel(tolower(age), c("young", "old"))) %>%
ggplot(aes(x = nuclear, y = prob, fill = mito)) +
geom_line(aes(group = mito, colour = mito), position = position_dodge(width = .5)) +
# geom_errorbar(aes(ymin = asymp.LCL, ymax = asymp.UCL),
# width = .25, position = position_dodge(width = .5)) +
geom_point(size = 3, pch = 21, position = position_dodge(width = .5)) +
labs(y = 'Proportion fertile (EMM ± 95% CI)') +
scale_colour_manual(values = met3) +
scale_fill_manual(values = met3) +
facet_wrap(~age) +
theme_bw() +
theme() +
NULL
comb_fert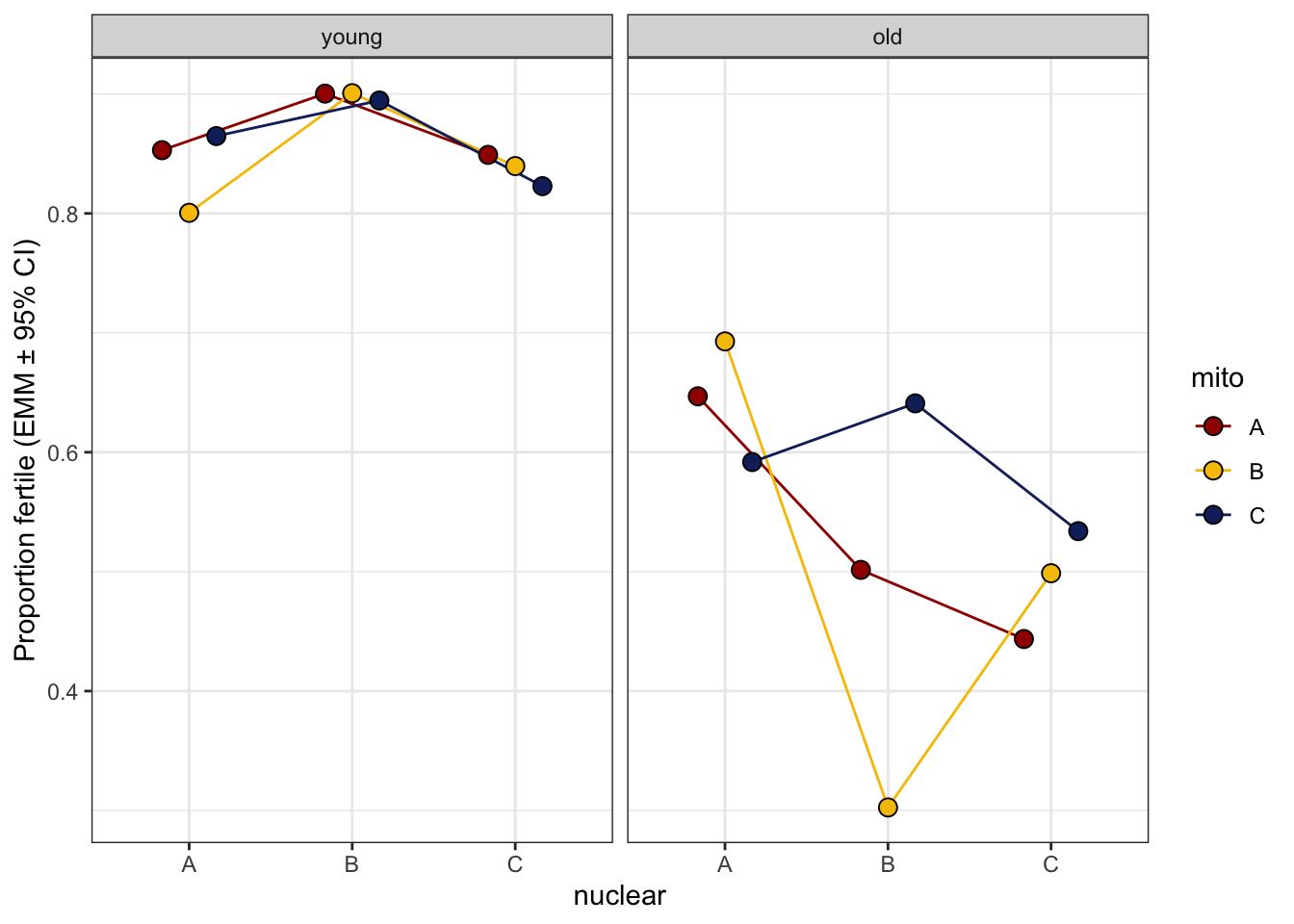
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
> Sperm viability
We measured sperm viability using live/dead staining in a control
medium (t0 = c) and after 30mins in a stressor medium (t30
= t). First, we analysed control and treatment separately.
For the measurement of sperm viability, we collected all males on a
single day for each block. However, the dissection,
staining and incubation protocol necessitated measurements were carried
out over several days. The measurement of sperm viability in young males
(collected at 5 days old) thus extended for 8 days and that of old males
(collected at 6 weeks old) for 6 days. To account for this variation in
age we included dissection day (an extension of male age)
and block as fixed covariates.
The sperm viability models returned a singularity warning message due to a zero-variance component estimate for the line random effect intercept. We retained these models after trying a range of alternatives and checking simplified models without the random slopes term that all resulted in the same qualitative findings for the fixed effects part of the model.
sperm_vib_filt %>%
group_by(mito, nuclear, age, treatment) %>%
summarise(mn = mean(viability),
se = sd(viability)/sqrt(n()),
s95 = se * 1.96) %>%
ggplot(aes(x = nuclear, y = mn, fill = mito)) +
geom_point(data = sperm_vib_filt %>%
group_by(mito, nuclear, age, treatment, male_ID) %>%
summarise(viability = mean(viability)),
aes(y = viability, colour = mito), alpha = .15,
position = position_jitterdodge(dodge.width = .5, jitter.width = .1)) +
geom_errorbar(aes(ymin = mn - s95, ymax = mn + s95),
width = .25, position = position_dodge(width = .5)) +
geom_line(aes(group = mito), position = position_dodge(width = .5)) +
geom_point(size = 3, pch = 21, position = position_dodge(width = .5)) +
labs(y = 'Sperm viability') +
facet_grid(treatment ~ age, scales = 'free_x') +
scale_colour_manual(values = met3) +
scale_fill_manual(values = met3) +
theme_bw() +
theme() +
NULL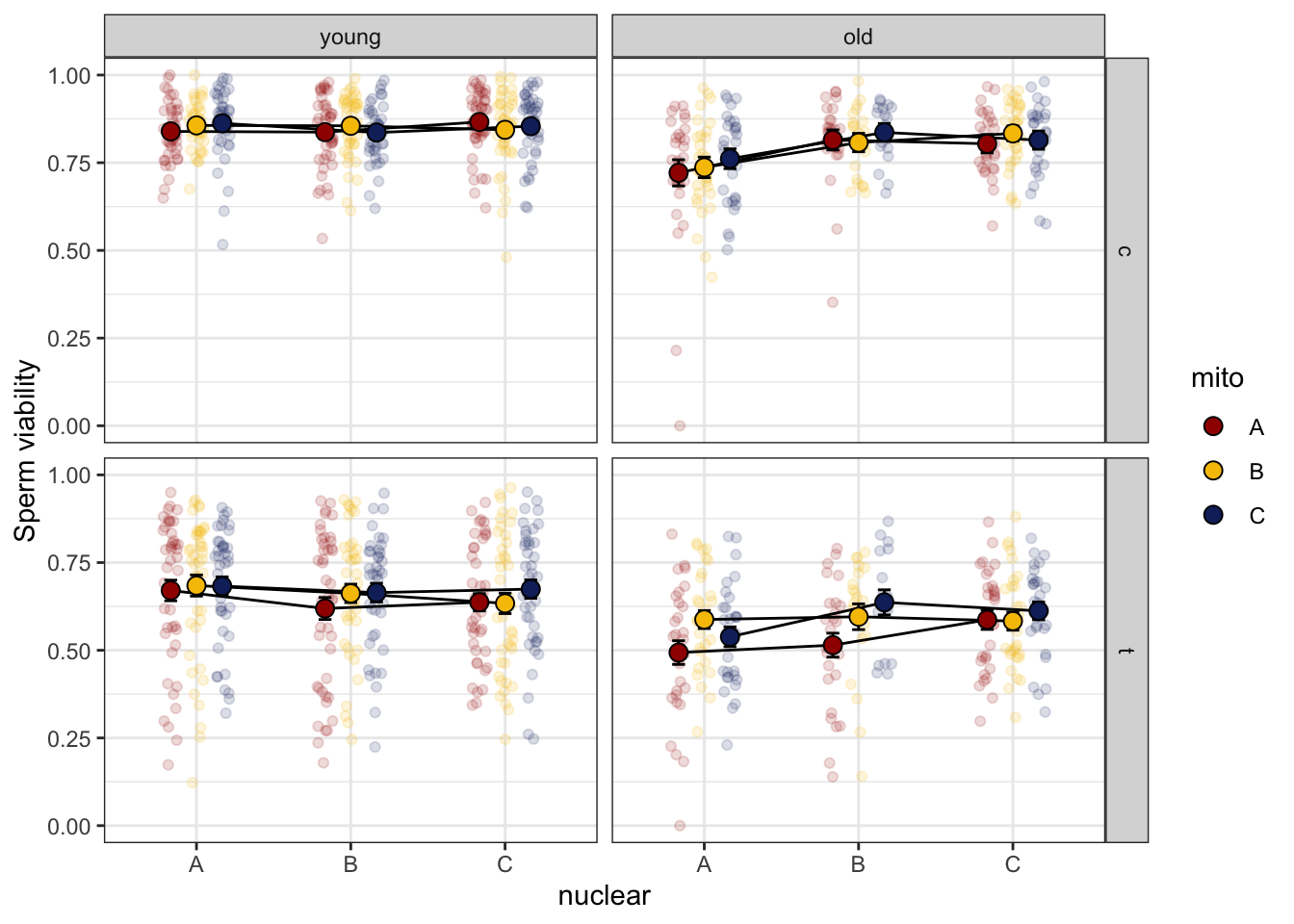
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
>>> Control treatment (t0)
First we take a look at the data to see how response changes with the covariates.
>>>> Day effect
spvfilt_control <- sperm_vib_filt %>% filter(treatment == 'c')
spvfilt_control %>%
group_by(mito, nuclear, age, days) %>%
summarise(mn = mean(viability),
se = sd(viability)/sqrt(n()),
s95 = se * 1.96) %>%
ggplot(aes(x = nuclear, y = mn, fill = mito)) +
geom_point(data = spvfilt_control %>%
group_by(mito, nuclear, age, days, male_ID) %>% summarise(viability = mean(viability)),
aes(y = viability, colour = mito), alpha = .15,
position = position_jitterdodge(dodge.width = .5, jitter.width = .1)) +
geom_errorbar(aes(ymin = mn - s95, ymax = mn + s95),
width = .25, position = position_dodge(width = .5)) +
geom_line(aes(group = mito, colour = mito), position = position_dodge(width = .5)) +
geom_point(size = 3, pch = 21, position = position_dodge(width = .5)) +
labs(y = 'Sperm viability') +
facet_grid(age ~ days, scales = 'free_x') +
scale_colour_manual(values = met3) +
scale_fill_manual(values = met3) +
theme_bw() +
theme() +
NULL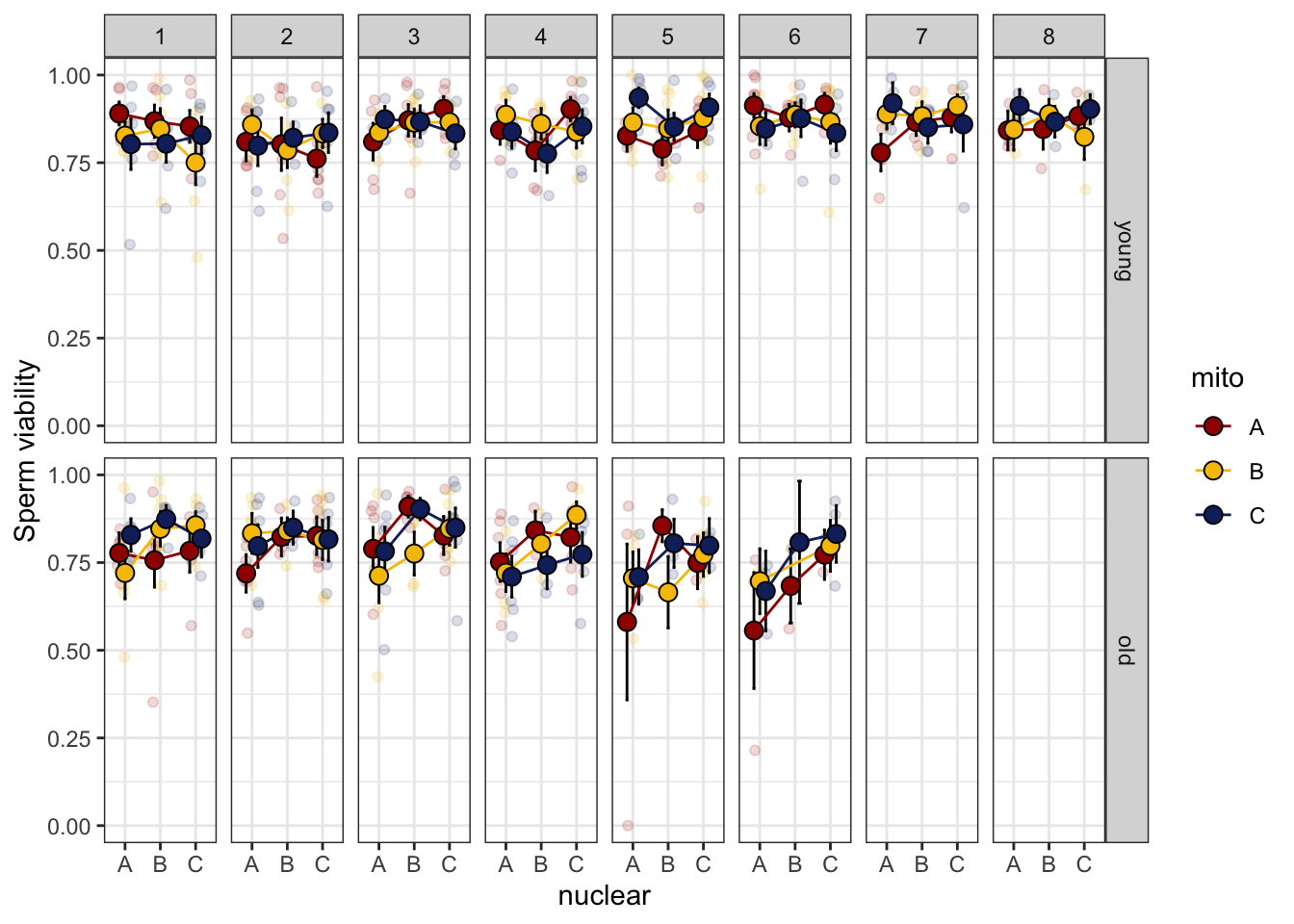
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
>>>> Block effect
spvfilt_control %>%
group_by(mito, nuclear, age, block) %>%
summarise(mn = mean(viability),
se = sd(viability)/sqrt(n()),
s95 = se * 1.96) %>%
ggplot(aes(x = nuclear, y = mn, fill = mito)) +
geom_point(data = spvfilt_control %>%
group_by(mito, nuclear, age, block, male_ID) %>% summarise(viability = mean(viability)),
aes(y = viability, colour = mito), alpha = .15,
position = position_jitterdodge(dodge.width = .5, jitter.width = .1)) +
geom_errorbar(aes(ymin = mn - s95, ymax = mn + s95),
width = .25, position = position_dodge(width = .5)) +
geom_line(aes(group = mito, colour = mito), position = position_dodge(width = .5)) +
geom_point(size = 3, pch = 21, position = position_dodge(width = .5)) +
labs(y = 'Sperm viability') +
facet_grid(age ~ block, scales = 'free_x') +
scale_colour_manual(values = met3) +
scale_fill_manual(values = met3) +
theme_bw() +
theme() +
NULL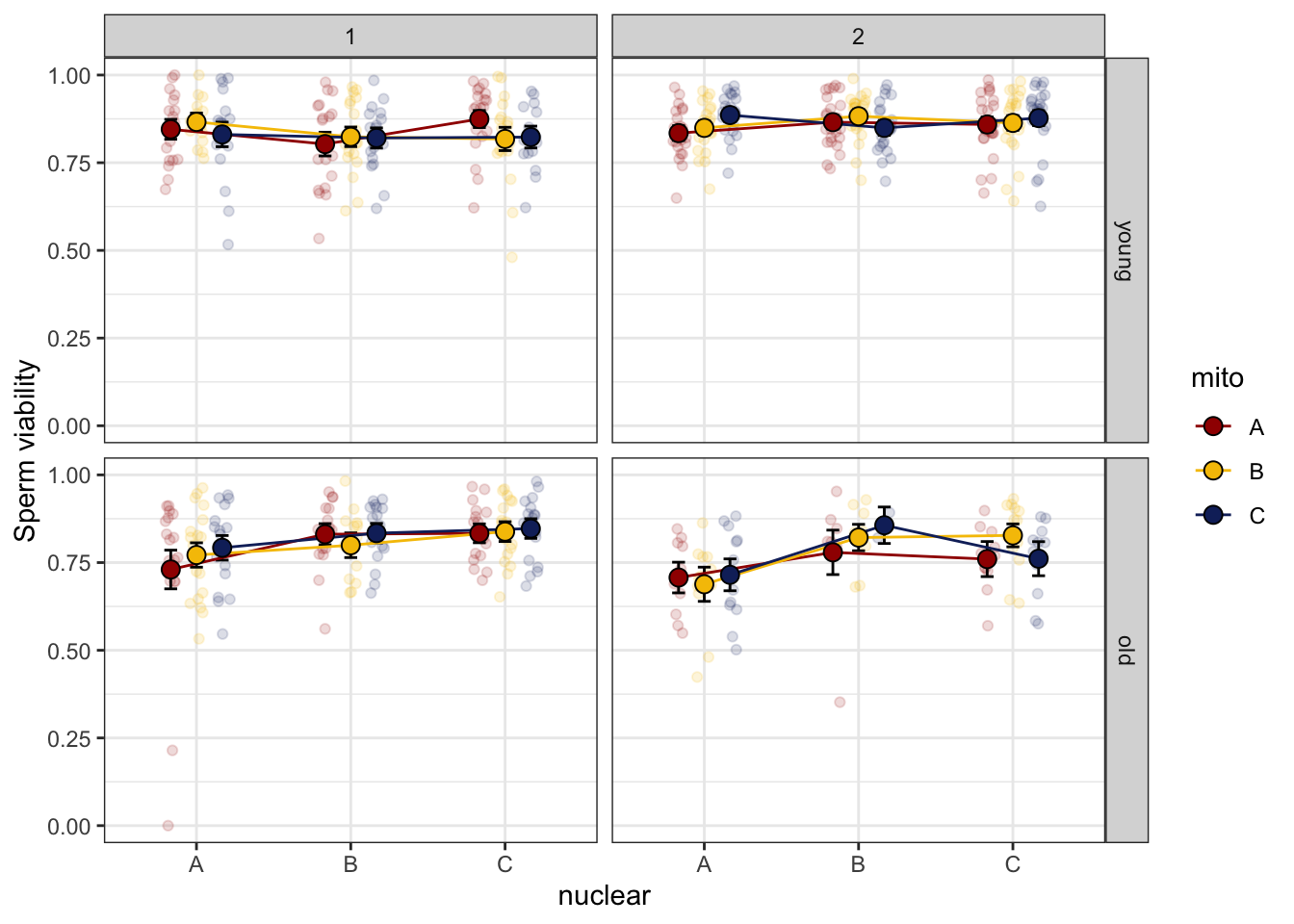
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
>>> Results
viability_m1 <- glmer(cbind(live, dead) ~ mito * nuclear * age + days + block + (1|LINE:age) + (1|male_ID) + (1|OLRE),
data = spvfilt_control, family = 'binomial',
control = glmerControl(optimizer = "bobyqa", optCtrl = list(maxfun = 50000)))>>>> Check diagnostics
performance::check_model(viability_m1)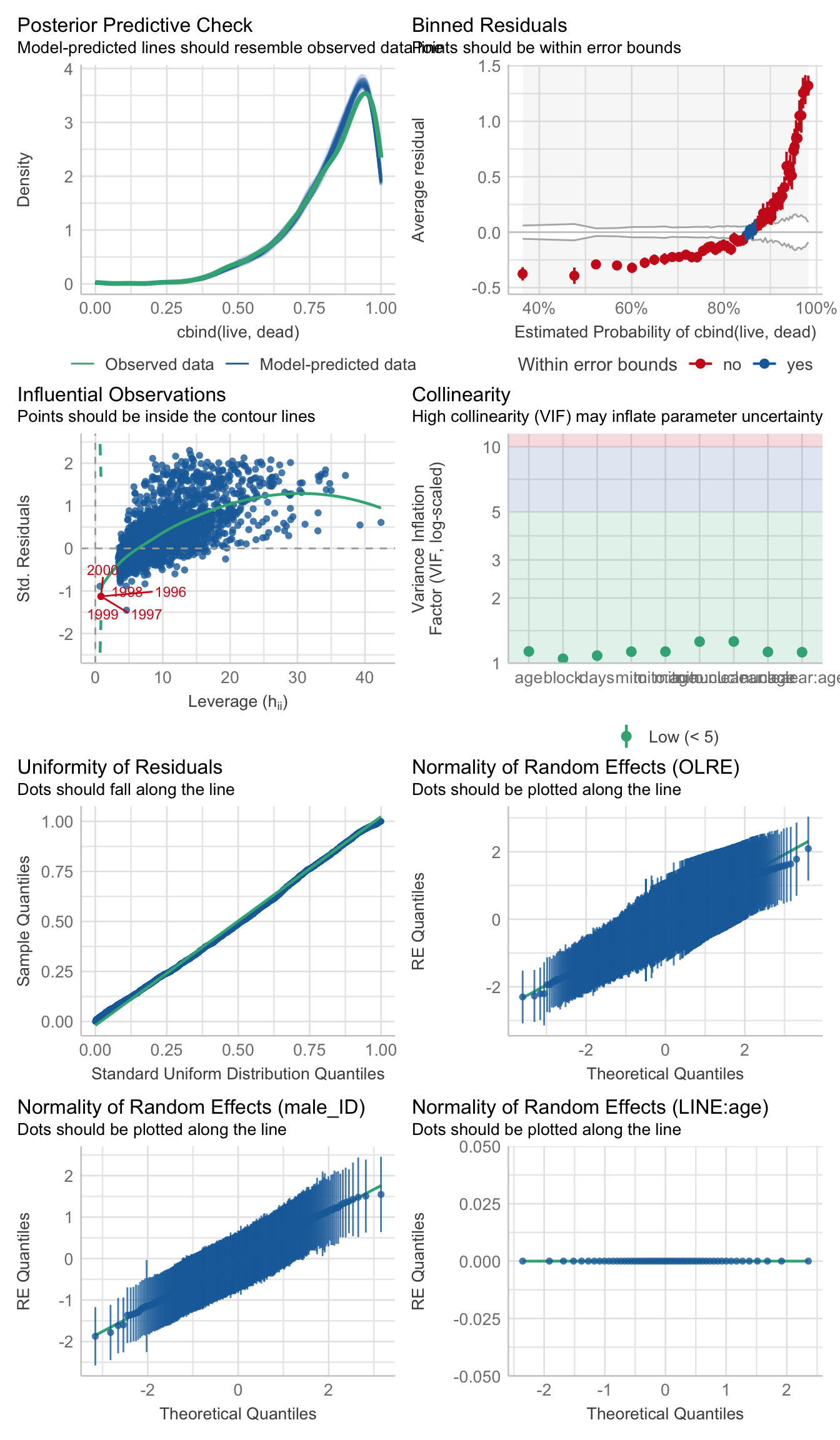
performance::check_overdispersion(viability_m1)# Overdispersion test
dispersion ratio = 0.625
p-value = < 0.001
# diagnostics
testDispersion(viability_m1)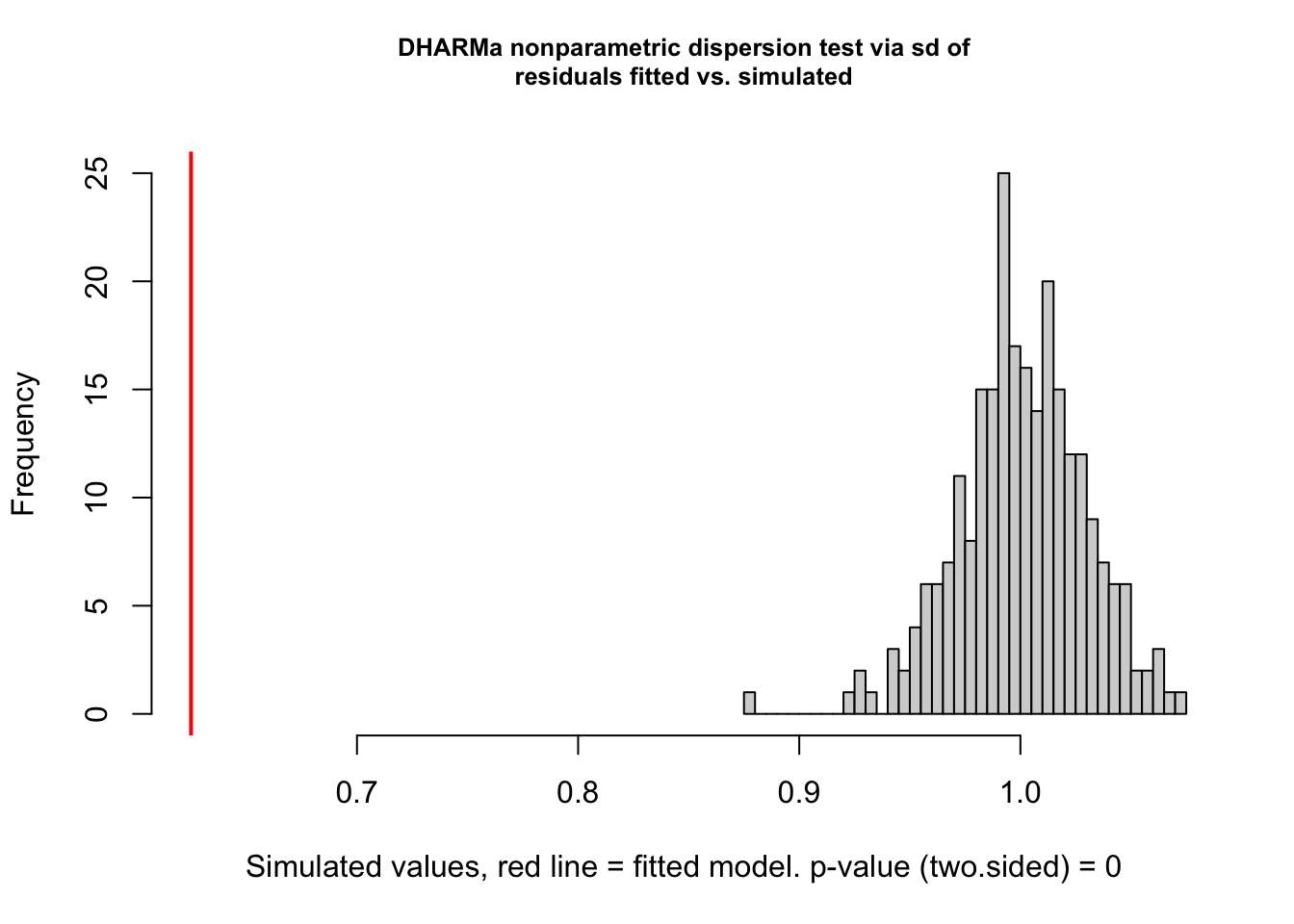
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
DHARMa nonparametric dispersion test via sd of residuals fitted vs.
simulated
data: simulationOutput
dispersion = 0.62462, p-value < 2.2e-16
alternative hypothesis: two.sided
simulationOutput <- simulateResiduals(fittedModel = viability_m1, plot = FALSE)
hist(residuals(simulationOutput))
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
hist(residuals(simulationOutput, quantileFunction = qnorm, outlierValues = c(-7,7)))
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
plot(simulationOutput)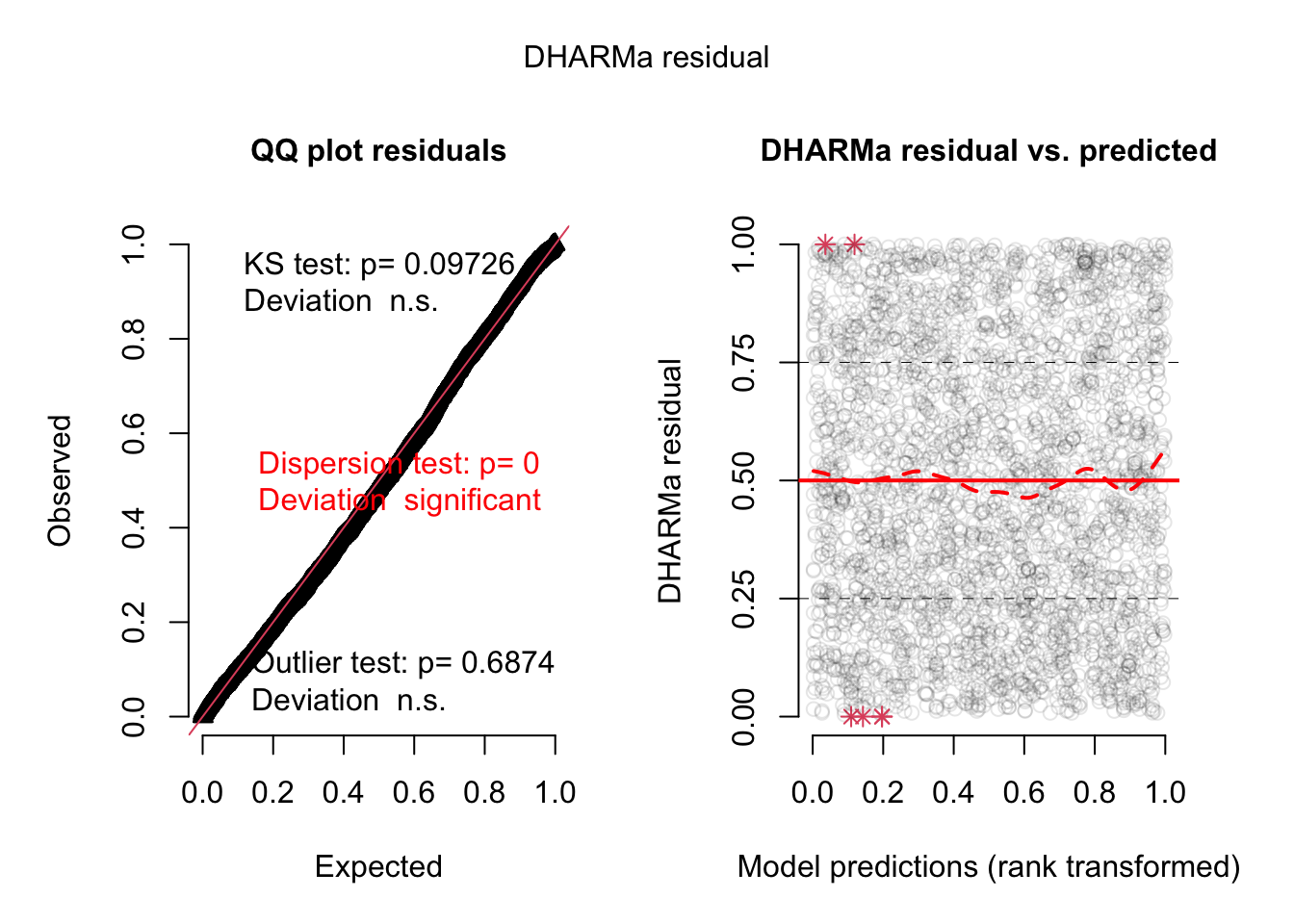
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
>>>>
car::Anova(viability_m1, type = "3") %>%
broom::tidy() %>%
#write_csv("output/anova_tables/viability_t0.csv") %>% # save anova table for supp. tables
as_tibble() %>%
kable(digits = 3,
caption = 'Analysis of Deviance Table (Type III Wald chisquare tests)') %>%
kable_styling(full_width = FALSE)| term | statistic | df | p.value |
|---|---|---|---|
| (Intercept) | 241.616 | 1 | 0.000 |
| mito | 0.986 | 2 | 0.611 |
| nuclear | 13.078 | 2 | 0.001 |
| age | 34.305 | 1 | 0.000 |
| days | 2.467 | 1 | 0.116 |
| block | 0.389 | 1 | 0.533 |
| mito:nuclear | 1.755 | 4 | 0.781 |
| mito:age | 0.125 | 2 | 0.939 |
| nuclear:age | 13.100 | 2 | 0.001 |
| mito:nuclear:age | 3.910 | 4 | 0.418 |
#summary(viability_m1)
# posthoc tests
bind_rows(emmeans(viability_m1, pairwise ~ age | nuclear, adjust = "tukey")$contrasts %>% broom::tidy(),
emmeans(viability_m1, pairwise ~ nuclear | age, adjust = "tukey")$contrasts %>% broom::tidy() %>%
rename(p.value = adj.p.value)) %>%
mutate(p.val = ifelse(p.value < 0.001, '< 0.001', round(p.value, 3))) %>%
select(-p.value, -term) %>% relocate(age, .before = contrast) %>%
kable(digits = 3,
caption = 'Posthoc Tukey tests to compare which groups differ') %>%
kable_styling(full_width = FALSE) %>%
kableExtra::group_rows("Age", 1, 3) %>%
kableExtra::group_rows("Nuclear - old", 4, 6) %>%
kableExtra::group_rows("Nuclear - young", 7, 9)| nuclear | age | contrast | null.value | estimate | std.error | df | statistic | p.val |
|---|---|---|---|---|---|---|---|---|
| Age | ||||||||
| A | NA | young - old | 0 | 0.750 | 0.114 | Inf | 6.574 | < 0.001 |
| B | NA | young - old | 0 | 0.179 | 0.128 | Inf | 1.397 | 0.163 |
| C | NA | young - old | 0 | 0.319 | 0.114 | Inf | 2.797 | 0.005 |
| Nuclear - old | ||||||||
| NA | young | A - B | 0 | 0.095 | 0.104 | Inf | 0.919 | 0.628 |
| NA | young | A - C | 0 | -0.067 | 0.104 | Inf | -0.647 | 0.794 |
| NA | young | B - C | 0 | -0.162 | 0.103 | Inf | -1.574 | 0.257 |
| Nuclear - young | ||||||||
| NA | old | A - B | 0 | -0.476 | 0.133 | Inf | -3.582 | < 0.001 |
| NA | old | A - C | 0 | -0.499 | 0.121 | Inf | -4.134 | < 0.001 |
| NA | old | B - C | 0 | -0.023 | 0.133 | Inf | -0.170 | 0.984 |
>>>> Reaction norms
sperm_vib_norm <- emmeans(viability_m1, ~ mito * nuclear * age, type = 'response') %>% as_tibble() %>%
ggplot(aes(x = nuclear, y = prob, fill = mito)) +
geom_line(aes(group = mito, colour = mito), position = position_dodge(width = .5)) +
# geom_errorbar(aes(ymin = asymp.LCL, ymax = asymp.UCL),
# width = .25, position = position_dodge(width = .5)) +
geom_point(size = 3, pch = 21, position = position_dodge(width = .5)) +
labs(y = 'Sperm viability (EMM)') +
scale_colour_manual(values = met3) +
scale_fill_manual(values = met3) +
#scale_y_continuous(limits = c(0, 1.05)) +
facet_wrap(~ age) +
theme_bw() +
theme() +
NULL
sperm_vib_norm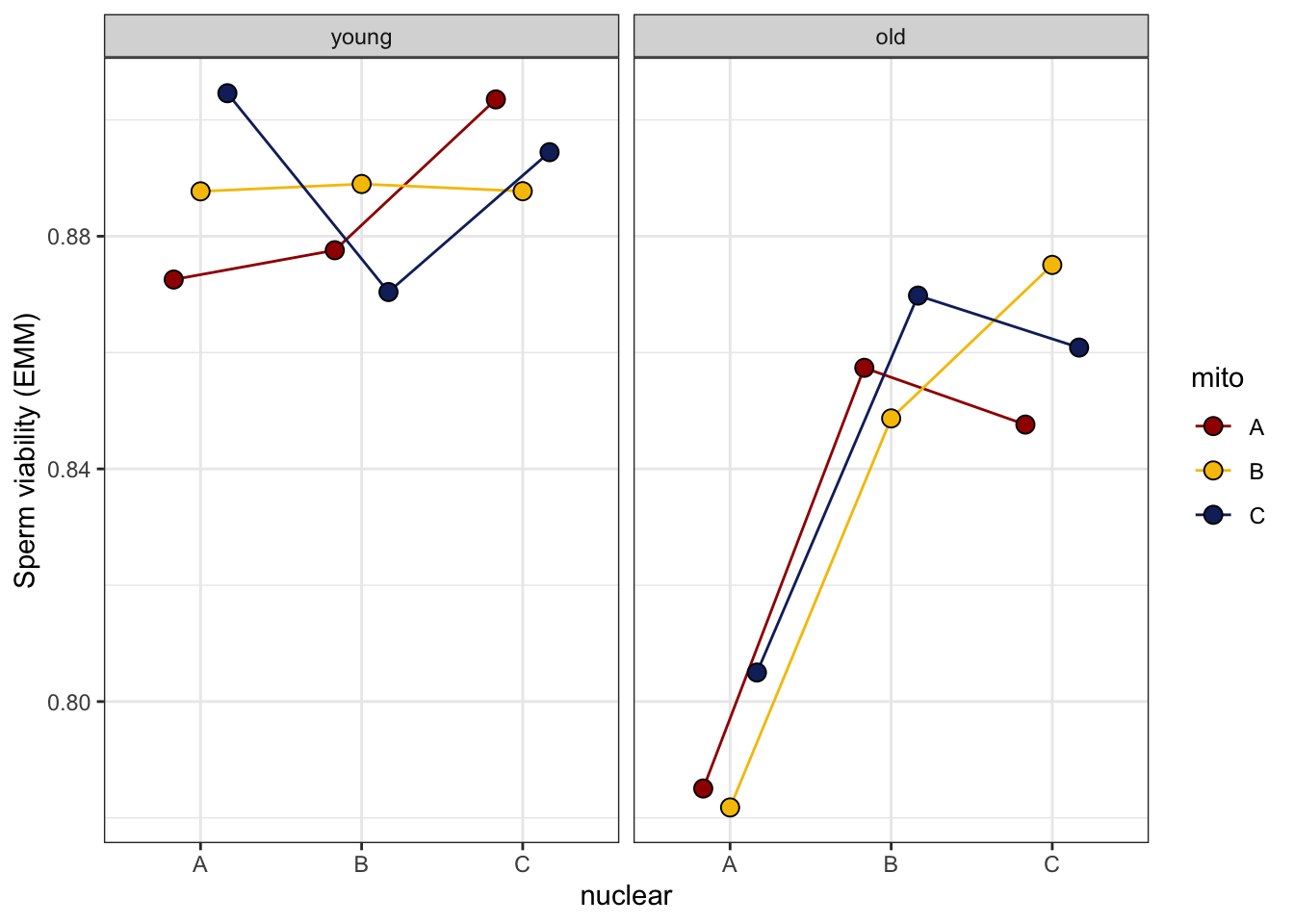
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
>>>> Raw data and means
sperm_vib_raw <- emmeans(viability_m1, ~ mito * nuclear * age, type = 'response') %>% as_tibble() %>%
ggplot(aes(x = nuclear, y = prob, fill = mito)) +
geom_jitter(data = spvfilt_control %>%
group_by(mito, nuclear, age, treatment, male_ID) %>%
summarise(viability = mean(viability)),
aes(y = viability, colour = mito),
position = position_jitterdodge(dodge.width = .5, jitter.width = .1),
size = 0.75, alpha = .1) +
geom_jitter(data = spvfilt_control %>%
group_by(LINE, age) %>%
summarise(mn = mean(viability)) %>%
separate(LINE, into = c("mito", "nuclear", NA), sep = "(?<=.)", remove = FALSE),
aes(y = mn, colour = mito),
position = position_jitterdodge(dodge.width = .5, jitter.width = .1),
alpha = .85, size = 2) +
geom_errorbar(aes(ymin = asymp.LCL, ymax = asymp.UCL),
width = .25, position = position_dodge(width = .5)) +
geom_point(size = 3, pch = 21, position = position_dodge(width = .5)) +
labs(y = 'Sperm viability (EMM ± 95% CIs)') +
scale_colour_manual(values = met3) +
scale_fill_manual(values = met3) +
facet_wrap(~ age) +
theme_bw() +
theme() +
geom_text(data = spvfilt_control %>%
group_by(mito, nuclear, age) %>%
summarise(N = n_distinct(male_ID)),
aes(y = -0.04, label = N),
size = 2, position = position_dodge(width = .5)) +
#ggsave('figures/sperm_vib_contrl_supp.pdf', height = 4, width = 8, dpi = 600, useDingbats = FALSE) +
NULL
sperm_vib_raw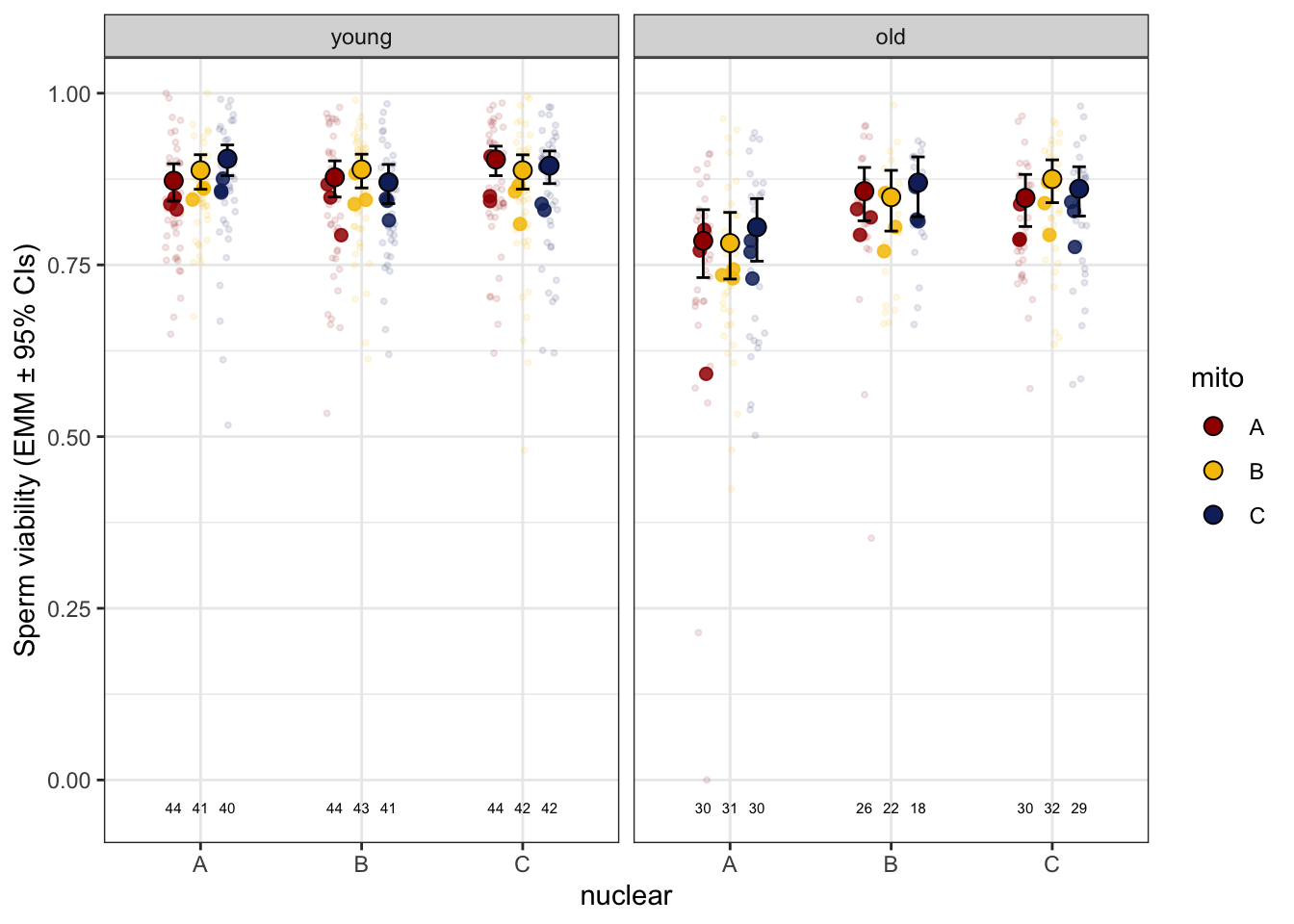
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
Matched vs. mismatched
viability_coevo <- glmer(cbind(live, dead) ~ coevolved * age + days + block + (age|LINE) + (1|male_ID) + (1|OLRE),
data = spvfilt_control, family = 'binomial')
car::Anova(viability_coevo, type = "3")Analysis of Deviance Table (Type III Wald chisquare tests)
Response: cbind(live, dead)
Chisq Df Pr(>Chisq)
(Intercept) 222.9440 1 < 2.2e-16 ***
coevolved 0.2777 1 0.5982
age 28.5398 1 9.179e-08 ***
days 2.3157 1 0.1281
block 0.4828 1 0.4872
coevolved:age 0.0162 1 0.8988
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Mito-type analysis
viability_snp <- glmer(cbind(live, dead) ~ mito_snp * nuclear * age +
days + block + (1 + age|LINE) + (1|male_ID) + (1|OLRE),
data = spvfilt_control, family = 'binomial',
control= glmerControl(optimizer="bobyqa", optCtrl=list(maxfun=50000)))
car::Anova(viability_snp, type = "3")Analysis of Deviance Table (Type III Wald chisquare tests)
Response: cbind(live, dead)
Chisq Df Pr(>Chisq)
(Intercept) 94.0598 1 < 2.2e-16 ***
mito_snp 6.2862 8 0.615205
nuclear 0.2596 2 0.878279
age 6.9185 1 0.008531 **
days 2.5477 1 0.110455
block 0.4244 1 0.514727
mito_snp:nuclear 6.5456 7 0.477669
mito_snp:age 3.1756 8 0.922859
nuclear:age 4.1995 2 0.122486
mito_snp:nuclear:age 5.3770 7 0.614063
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
>> Stress treatment (t30)
spvfilt_treat <- sperm_vib_filt %>% filter(treatment == 't')>>>> Day effect
spvfilt_treat %>%
group_by(mito, nuclear, age, block) %>%
summarise(mn = mean(viability),
se = sd(viability)/sqrt(n()),
s95 = se * 1.96) %>%
ggplot(aes(x = nuclear, y = mn, fill = mito)) +
geom_point(data = spvfilt_treat %>%
group_by(mito, nuclear, age, block, male_ID) %>% summarise(viability = mean(viability)),
aes(y = viability, colour = mito), alpha = .15,
position = position_jitterdodge(dodge.width = .5, jitter.width = .1)) +
geom_errorbar(aes(ymin = mn - s95, ymax = mn + s95),
width = .25, position = position_dodge(width = .5)) +
geom_line(aes(group = mito), position = position_dodge(width = .5)) +
geom_point(size = 3, pch = 21, position = position_dodge(width = .5)) +
labs(y = 'Sperm viability') +
facet_grid(age ~ block, scales = 'free_x') +
scale_colour_manual(values = met3) +
scale_fill_manual(values = met3) +
theme_bw() +
theme() +
NULL
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
>>>> Block effect
spvfilt_treat %>%
group_by(mito, nuclear, age, days) %>%
summarise(mn = mean(viability),
se = sd(viability)/sqrt(n()),
s95 = se * 1.96) %>%
ggplot(aes(x = nuclear, y = mn, fill = mito)) +
geom_point(data = spvfilt_treat %>%
group_by(mito, nuclear, age, days, male_ID) %>% summarise(viability = mean(viability)),
aes(y = viability, colour = mito), alpha = .15,
position = position_jitterdodge(dodge.width = .5, jitter.width = .1)) +
geom_errorbar(aes(ymin = mn - s95, ymax = mn + s95),
width = .25, position = position_dodge(width = .5)) +
geom_line(aes(group = mito), position = position_dodge(width = .5)) +
geom_point(size = 3, pch = 21, position = position_dodge(width = .5)) +
labs(y = 'Sperm viability') +
facet_grid(age ~ days, scales = 'free_x') +
scale_colour_manual(values = met3) +
scale_fill_manual(values = met3) +
theme_bw() +
theme() +
NULL
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
>>> Results
# model
viability_m2 <- glmer(cbind(live, dead) ~ mito * nuclear * age + days + block + (1 + age|LINE) + (1|male_ID) + (1|OLRE),
data = spvfilt_treat, family = 'binomial',
control = glmerControl(optimizer = "bobyqa", optCtrl = list(maxfun = 50000)))>>>> Check diagnostics
performance::check_model(viability_m2)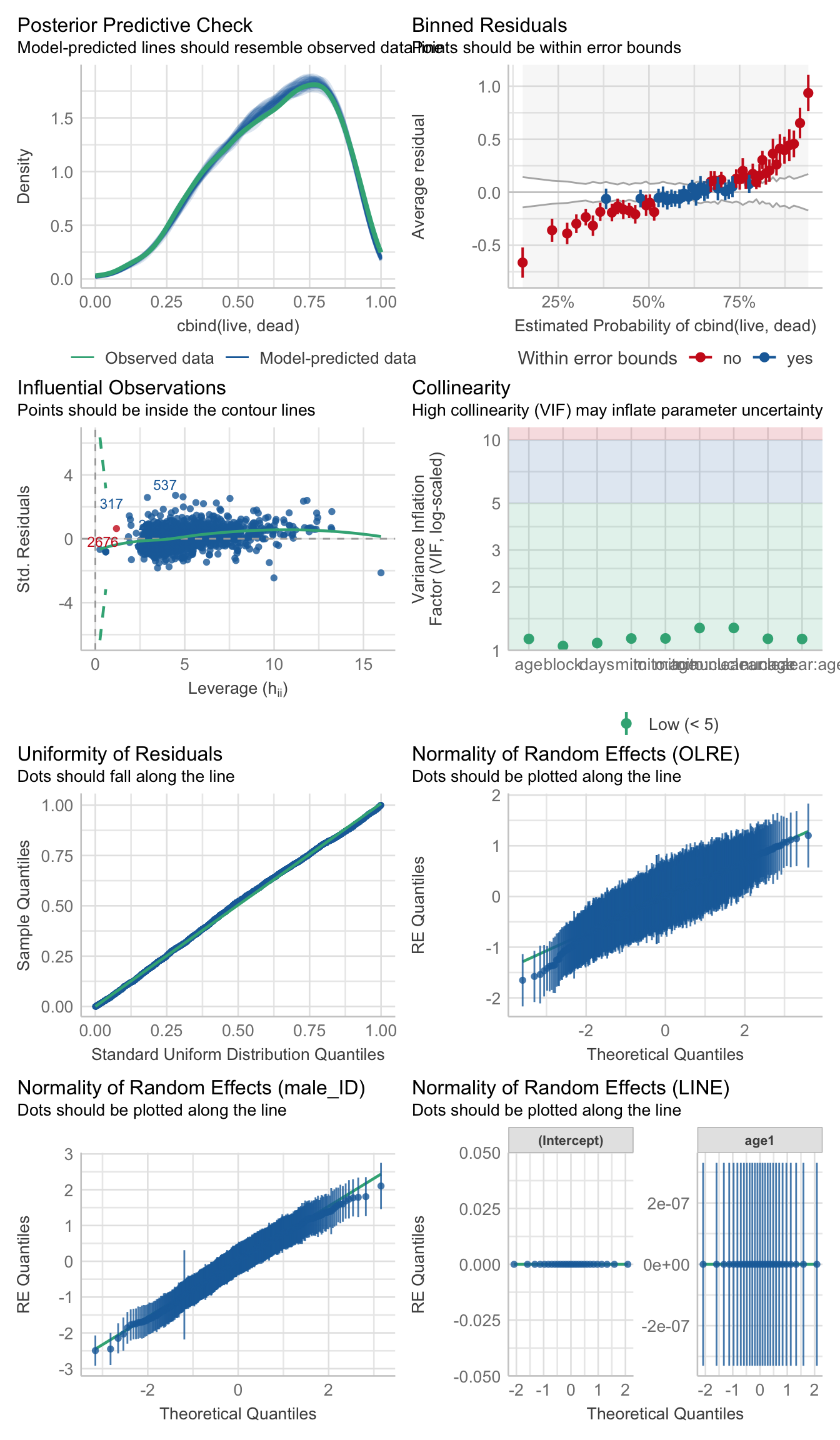
performance::check_overdispersion(viability_m2)# Overdispersion test
dispersion ratio = 0.854
p-value = < 0.001
testDispersion(viability_m2)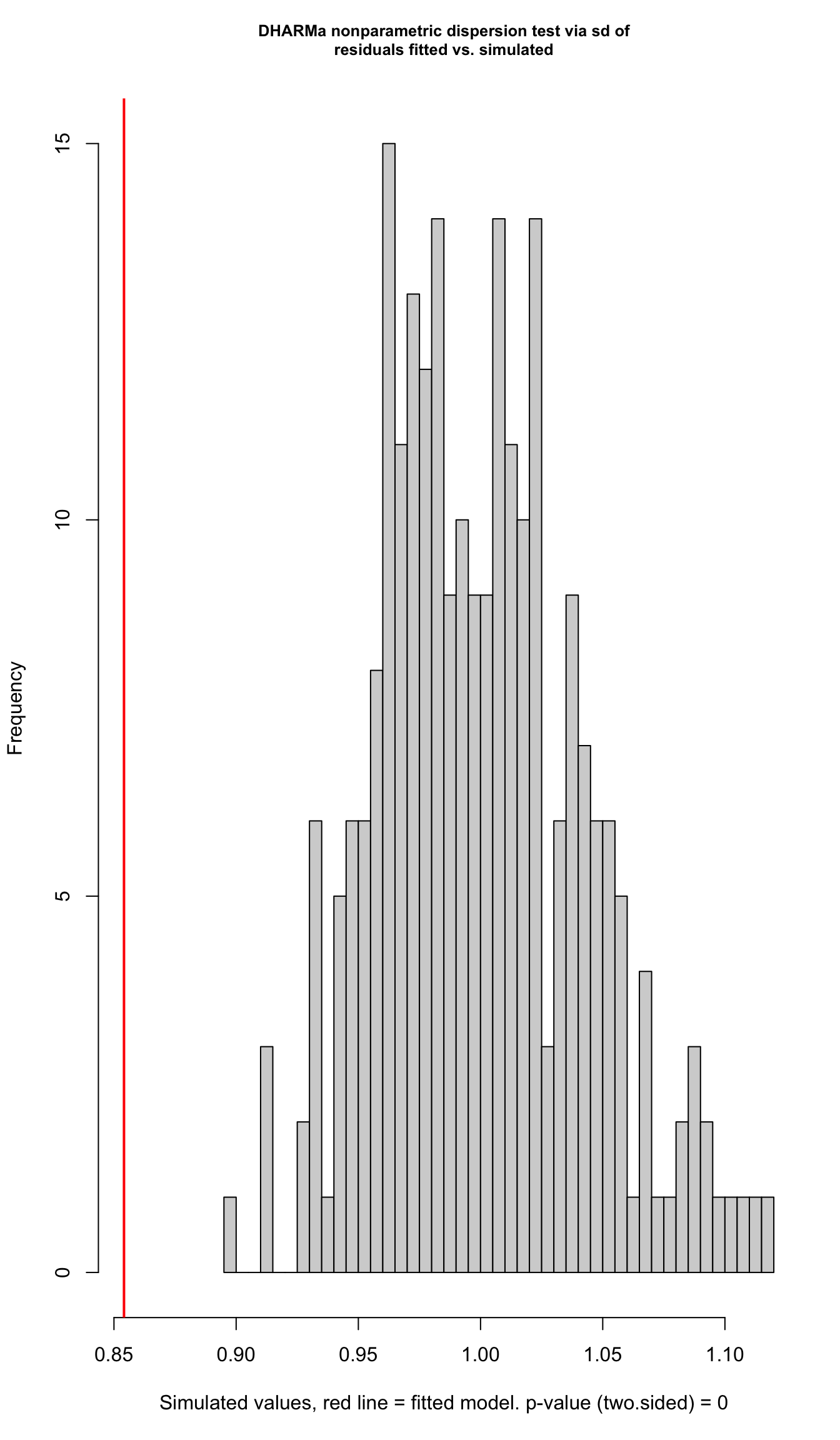
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
DHARMa nonparametric dispersion test via sd of residuals fitted vs.
simulated
data: simulationOutput
dispersion = 0.85413, p-value < 2.2e-16
alternative hypothesis: two.sided
simulationOutput <- simulateResiduals(fittedModel = viability_m2, plot = FALSE)
hist(residuals(simulationOutput))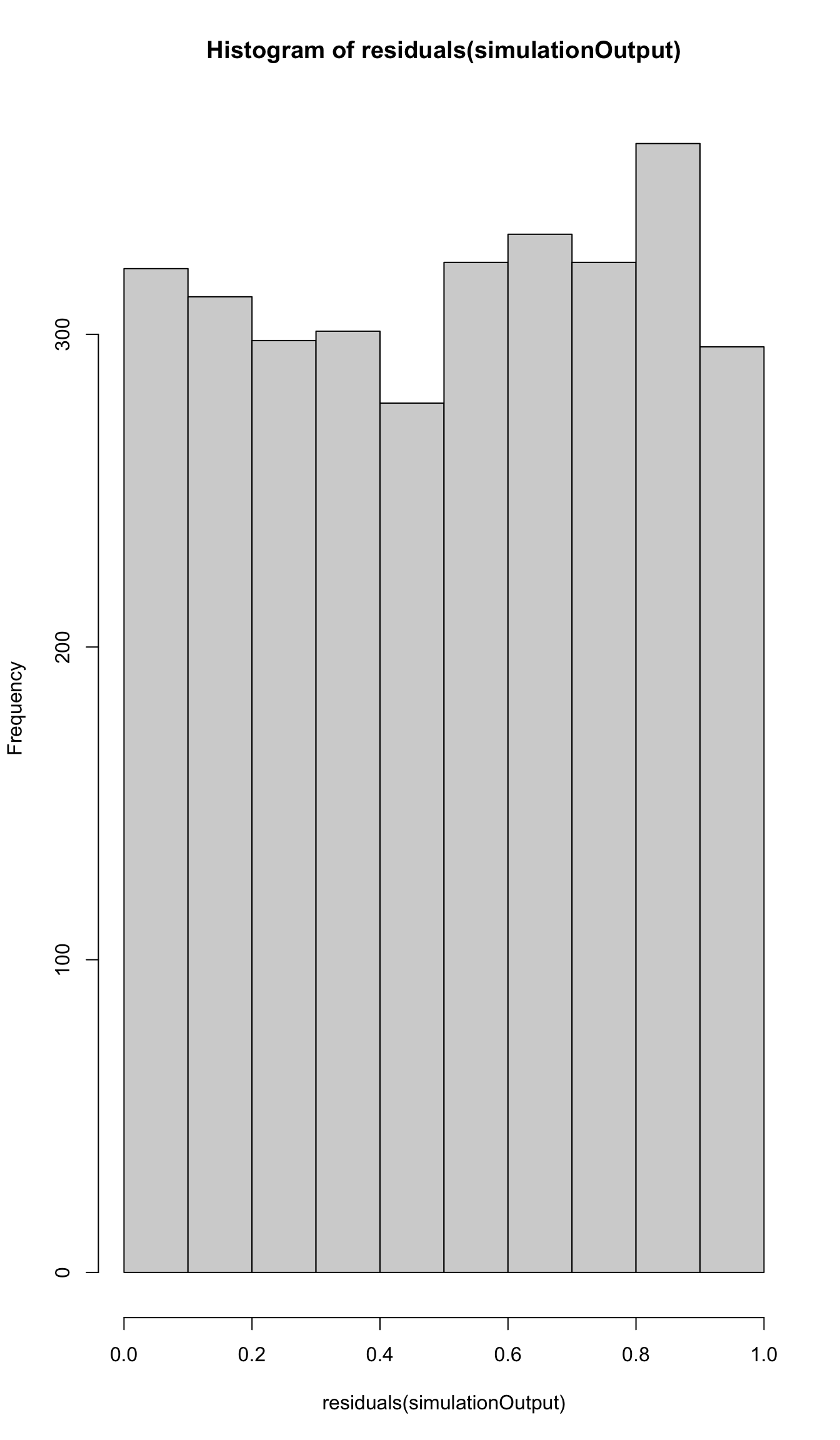
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
hist(residuals(simulationOutput, quantileFunction = qnorm, outlierValues = c(-7,7)))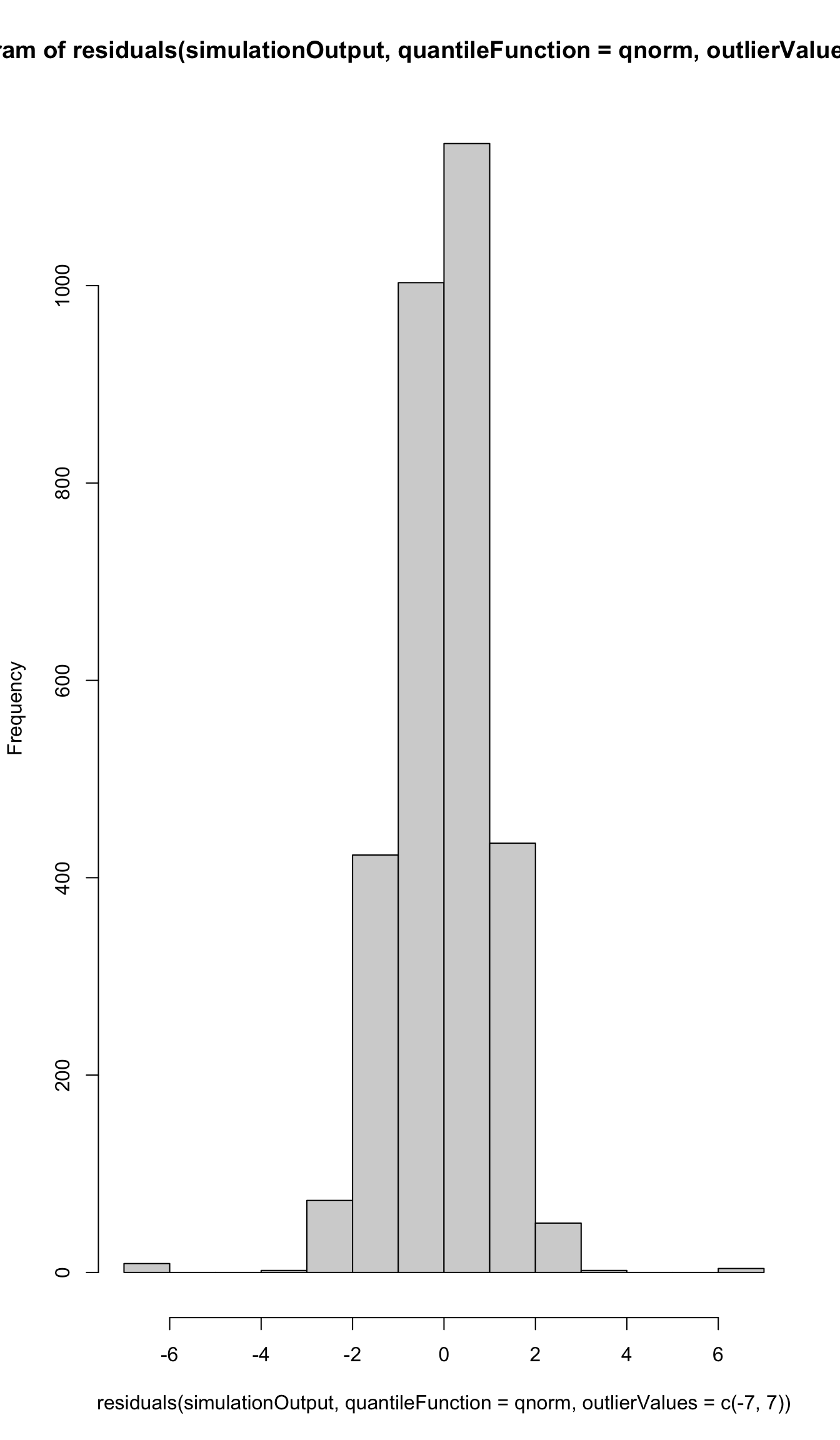
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
plot(simulationOutput)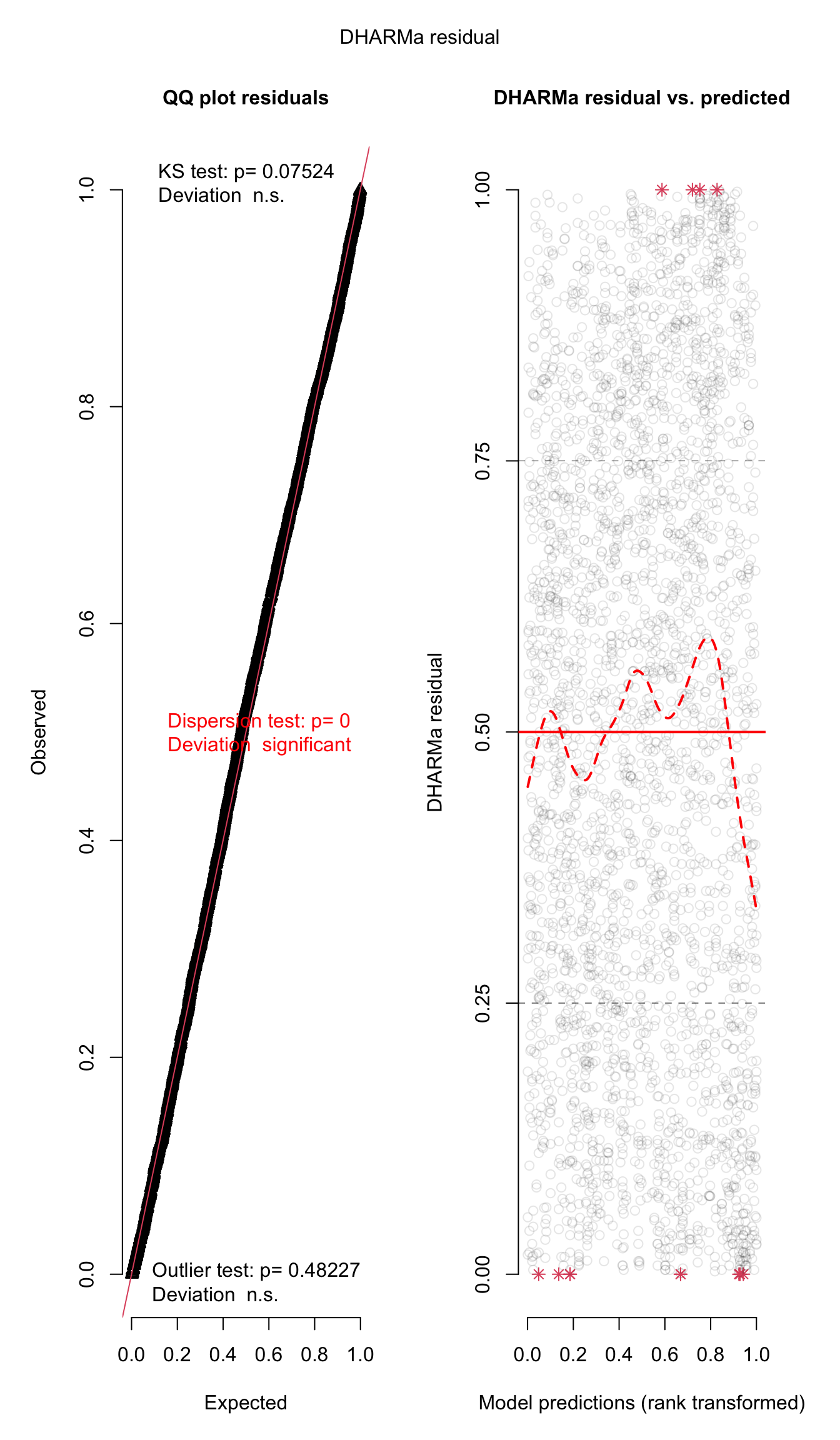
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
>>>> Results
car::Anova(viability_m2, type = "3") %>%
broom::tidy() %>%
#write_csv("output/anova_tables/viability_t30.csv") %>% # save anova table for supp. tables
as_tibble() %>%
kable(digits = 3,
caption = 'Analysis of Deviance Table (Type III Wald chisquare tests)') %>%
kable_styling(full_width = FALSE)| term | statistic | df | p.value |
|---|---|---|---|
| (Intercept) | 77.698 | 1 | 0.000 |
| mito | 7.119 | 2 | 0.028 |
| nuclear | 0.545 | 2 | 0.761 |
| age | 58.101 | 1 | 0.000 |
| days | 3.514 | 1 | 0.061 |
| block | 15.118 | 1 | 0.000 |
| mito:nuclear | 3.759 | 4 | 0.440 |
| mito:age | 0.853 | 2 | 0.653 |
| nuclear:age | 6.022 | 2 | 0.049 |
| mito:nuclear:age | 1.778 | 4 | 0.777 |
#summary(viability_m2)
# posthoc tests
bind_rows(emmeans(viability_m2, pairwise ~ mito, adjust = "tukey")$contrasts %>% broom::tidy() %>%
rename(p.value = adj.p.value),
emmeans(viability_m2, pairwise ~ block, adjust = "tukey")$contrasts %>% broom::tidy(),
emmeans(viability_m2, pairwise ~ age | nuclear, adjust = "tukey")$contrasts %>% broom::tidy(),
emmeans(viability_m2, pairwise ~ nuclear | age, adjust = "tukey")$contrasts %>% broom::tidy() %>%
rename(p.value = adj.p.value)) %>%
mutate(p.val = ifelse(p.value < 0.001, '< 0.001', round(p.value, 3))) %>%
select(-p.value, -term) %>% relocate(age, .before = contrast) %>%
kable(digits = 3,
caption = 'Posthoc Tukey tests to compare which groups differ') %>%
kable_styling(full_width = FALSE) %>%
kableExtra::group_rows("Mitochondria", 1, 3) %>%
kableExtra::group_rows("Block", 4, 4) %>%
kableExtra::group_rows("Age", 5, 7) %>%
kableExtra::group_rows("Nuclear - old", 8, 10) %>%
kableExtra::group_rows("Nuclear - young", 11, 13)| age | contrast | null.value | estimate | std.error | df | statistic | nuclear | p.val |
|---|---|---|---|---|---|---|---|---|
| Mitochondria | ||||||||
| NA | A - B | 0 | -0.182 | 0.084 | Inf | -2.158 | NA | 0.079 |
| NA | A - C | 0 | -0.208 | 0.086 | Inf | -2.412 | NA | 0.042 |
| NA | B - C | 0 | -0.026 | 0.087 | Inf | -0.297 | NA | 0.953 |
| Block | ||||||||
| NA | block1 - block2 | 0 | 0.270 | 0.069 | Inf | 3.888 | NA | < 0.001 |
| Age | ||||||||
| NA | young - old | 0 | 0.791 | 0.119 | Inf | 6.647 | A | < 0.001 |
| NA | young - old | 0 | 0.498 | 0.133 | Inf | 3.741 | B | < 0.001 |
| NA | young - old | 0 | 0.399 | 0.118 | Inf | 3.376 | C | < 0.001 |
| Nuclear - old | ||||||||
| young | A - B | 0 | 0.170 | 0.107 | Inf | 1.584 | NA | 0.253 |
| young | A - C | 0 | 0.156 | 0.107 | Inf | 1.461 | NA | 0.31 |
| young | B - C | 0 | -0.013 | 0.106 | Inf | -0.124 | NA | 0.992 |
| Nuclear - young | ||||||||
| old | A - B | 0 | -0.123 | 0.139 | Inf | -0.889 | NA | 0.647 |
| old | A - C | 0 | -0.236 | 0.126 | Inf | -1.874 | NA | 0.146 |
| old | B - C | 0 | -0.113 | 0.139 | Inf | -0.813 | NA | 0.695 |
>>>>> Reaction norms
sperm_stress_norm <- emmeans(viability_m2, ~ mito * nuclear * age, type = 'response') %>% as_tibble() %>%
ggplot(aes(x = nuclear, y = prob, fill = mito)) +
geom_line(aes(group = mito, colour = mito), position = position_dodge(width = .5)) +
# geom_errorbar(aes(ymin = asymp.LCL, ymax = asymp.UCL),
# width = .25, position = position_dodge(width = .5)) +
geom_point(size = 3, pch = 21, position = position_dodge(width = .5)) +
labs(y = 'Sperm viability (EMM)') +
scale_colour_manual(values = met3) +
scale_fill_manual(values = met3) +
facet_wrap(~ age) +
theme_bw() +
theme() +
NULL
sperm_stress_norm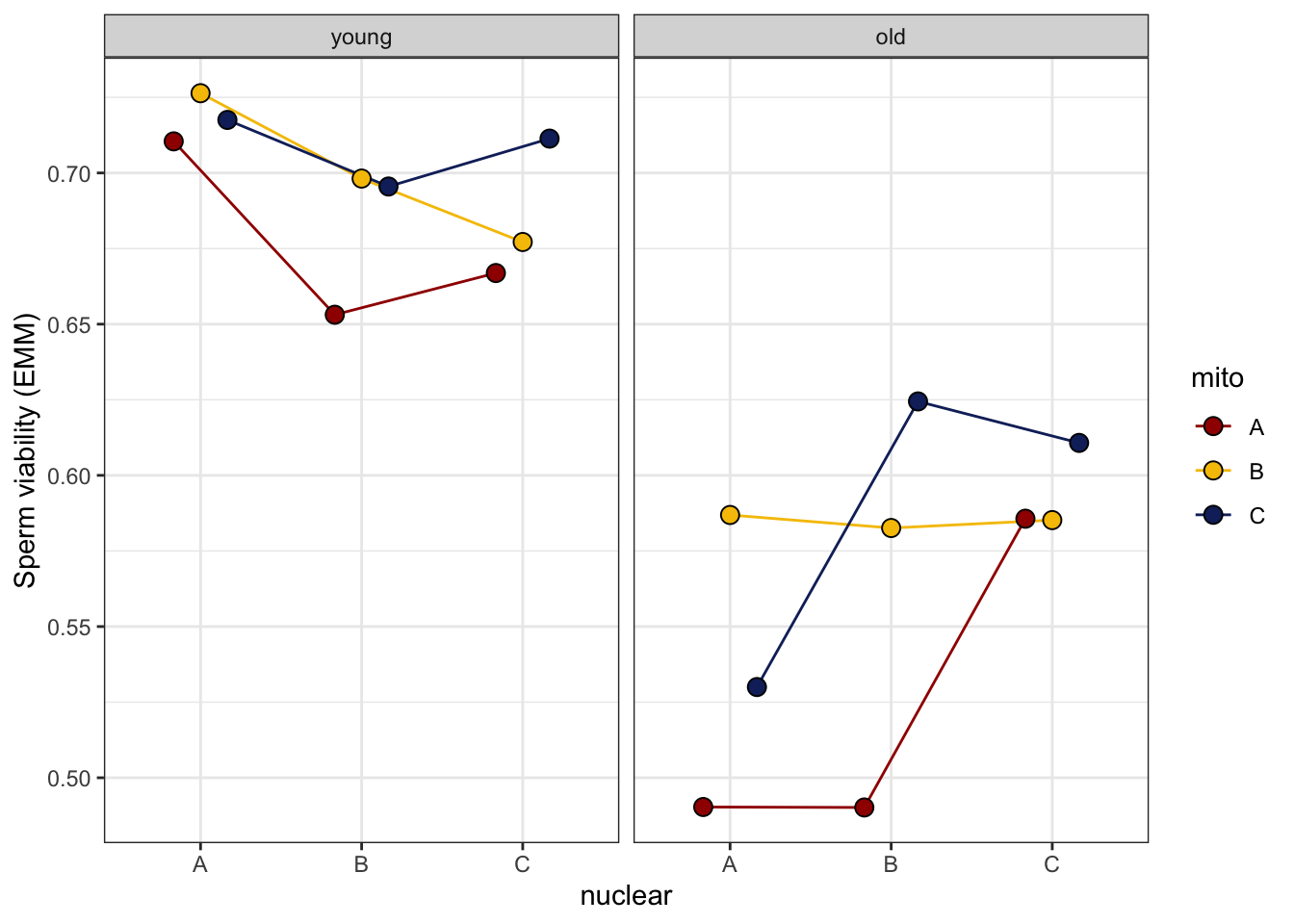
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
>>>>> Raw data and means
sperm_stress_raw <- emmeans(viability_m2, ~ mito * nuclear * age, type = 'response') %>% as_tibble() %>%
ggplot(aes(x = nuclear, y = prob, fill = mito)) +
geom_jitter(data = spvfilt_treat %>%
group_by(mito, nuclear, age, treatment, male_ID) %>%
summarise(viability = mean(viability)),
aes(y = viability, colour = mito),
position = position_jitterdodge(dodge.width = .5, jitter.width = .1),
size = 0.75, alpha = .1) +
geom_jitter(data = spvfilt_treat %>%
group_by(LINE, age) %>%
summarise(mn = mean(viability)) %>%
separate(LINE, into = c("mito", "nuclear", NA), sep = "(?<=.)", remove = FALSE),
aes(y = mn, colour = mito),
position = position_jitterdodge(dodge.width = .5, jitter.width = .1),
alpha = .85, size = 2) +
geom_errorbar(aes(ymin = asymp.LCL, ymax = asymp.UCL),
width = .25, position = position_dodge(width = .5)) +
geom_point(size = 3, pch = 21, position = position_dodge(width = .5)) +
labs(y = 'Sperm viability (EMM ± 95% CIs)') +
scale_colour_manual(values = met3) +
scale_fill_manual(values = met3) +
facet_wrap(~ age) +
theme_bw() +
theme() +
geom_text(data = spvfilt_control %>%
group_by(mito, nuclear, age) %>%
summarise(N = n_distinct(male_ID)),
aes(y = -0.04, label = N),
size = 2, position = position_dodge(width = .5)) +
#ggsave('figures/sperm_stress_raw.pdf', height = 4, width = 8, dpi = 600, useDingbats = FALSE) +
NULL
sperm_stress_raw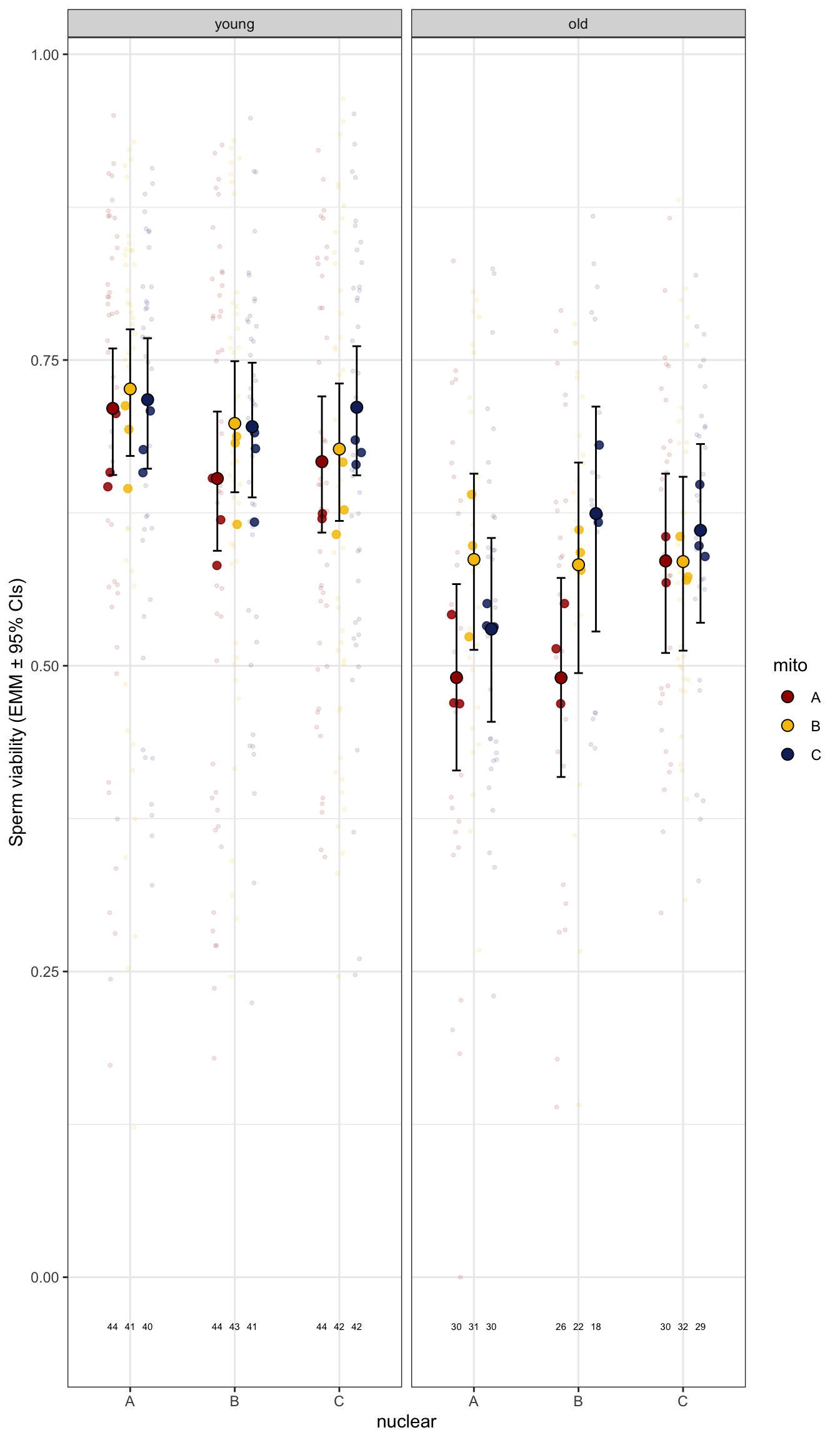
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
Matched vs. mismatched
stress_coevo <- glmer(cbind(live, dead) ~ coevolved * age + days + block + (age|LINE) + (1|male_ID) + (1|OLRE),
data = spvfilt_treat, family = 'binomial')
car::Anova(stress_coevo, type = "3")Analysis of Deviance Table (Type III Wald chisquare tests)
Response: cbind(live, dead)
Chisq Df Pr(>Chisq)
(Intercept) 75.6518 1 < 2.2e-16 ***
coevolved 0.1516 1 0.6970370
age 56.4580 1 5.741e-14 ***
days 3.5747 1 0.0586647 .
block 15.0434 1 0.0001051 ***
coevolved:age 0.5181 1 0.4716464
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Mito-type analysis
viability_m2_snp <- glmer(cbind(live, dead) ~ mito_snp * nuclear * age + days + block +
(1 + age|LINE) + (1|male_ID) + (1|OLRE),
data = spvfilt_treat, family = 'binomial',
control = glmerControl(optimizer = "bobyqa", optCtrl = list(maxfun = 50000)))
car::Anova(viability_m2_snp, type = "3")Analysis of Deviance Table (Type III Wald chisquare tests)
Response: cbind(live, dead)
Chisq Df Pr(>Chisq)
(Intercept) 28.9303 1 7.503e-08 ***
mito_snp 11.0643 8 0.19808
nuclear 0.5764 2 0.74962
age 5.6219 1 0.01774 *
days 3.6522 1 0.05600 .
block 15.2930 1 9.206e-05 ***
mito_snp:nuclear 2.0731 7 0.95568
mito_snp:age 0.9689 8 0.99844
nuclear:age 2.4954 2 0.28716
mito_snp:nuclear:age 2.8611 7 0.89755
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Combined plots
sperm_diffs_norm <- emmeans(viability_m1, ~ mito * nuclear * age, type = 'response') %>% as_tibble() %>%
ggplot(aes(x = nuclear, y = prob, fill = mito)) +
geom_line(aes(group = mito, colour = mito), position = position_dodge(width = .5)) +
# geom_errorbar(aes(ymin = asymp.LCL, ymax = asymp.UCL),
# width = .25, position = position_dodge(width = .5)) +
geom_point(size = 3, pch = 23, position = position_dodge(width = .5)) +
# after stress
geom_line(data = emmeans(viability_m2, ~ mito * nuclear * age, type = 'response') %>% as_tibble(),
aes(group = mito, colour = mito), position = position_dodge(width = .5)) +
# geom_errorbar(data = emmeans(viability_m2, ~ mito * nuclear * age, type = 'response') %>% as_tibble(),
# aes(ymin = asymp.LCL, ymax = asymp.UCL),
# width = .25, position = position_dodge(width = .5)) +
geom_point(data = emmeans(viability_m2, ~ mito * nuclear * age, type = 'response') %>% as_tibble(),
aes(fill = mito), size = 3, pch = 22, position = position_dodge(width = .5)) +
labs(y = 'Sperm viability (EMM)') +
scale_colour_manual(values = met3) +
scale_fill_manual(values = met3) +
facet_wrap(~ age) +
theme_bw() +
theme() +
NULL
sperm_diffs_norm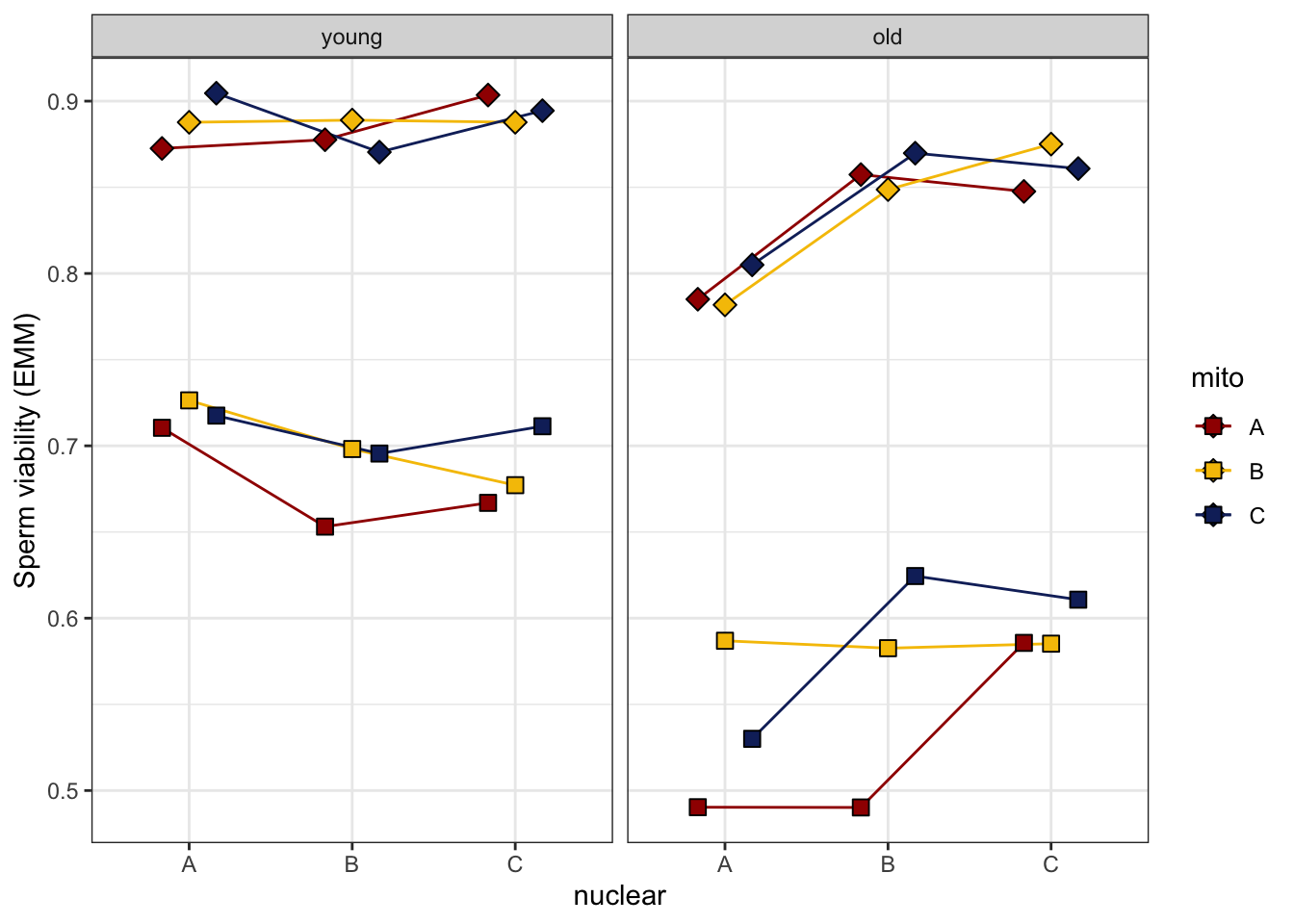
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
# bind_rows(
# emmeans(viability_m1, ~ mito * nuclear * age, type = 'response') %>% as_tibble() %>% mutate(stage = "t0"),
# emmeans(viability_m2, ~ mito * nuclear * age, type = 'response') %>% as_tibble() %>% mutate(stage = "t30")) %>%
# ggplot(aes(x = nuclear, y = prob, fill = mito, shape = stage)) +
# geom_errorbar(aes(ymin = asymp.LCL, ymax = asymp.UCL), position = position_dodge(width = .5), width = .5) +
# geom_line(aes(group = interaction(mito, stage), colour = mito), position = position_dodge(width = .5)) +
# geom_point(size = 3, position = position_dodge(width = .5)) +
# labs(y = 'No. progeny (EMM)') +
# scale_colour_manual(values = met3) +
# scale_fill_manual(values = met3) +
# scale_shape_manual(values = c(23, 22)) +
# facet_wrap(~ age) +
# theme_bw() +
# theme() +
# NULL>> Sperm quality
For the analysis of sperm quality we calculated the mean difference in sperm viability between treated and control for each male. In the plots below lines connect the mean measurements for each male, showing the overall average decline in viability after stress treatment.
# calculate difference in viability between treatment/control for each male
viability_diff <- sperm_vib_filt %>%
group_by(treatment, mito, mito_snp, nuclear, coevolved, LINE, age, male_ID, block, days) %>%
summarise(mn_vib = mean(viability)) %>% #filter(male_ID == 1) %>%
pivot_wider(names_from = treatment,
values_from = mn_vib) %>%
mutate(diff = t - c)>>> Young males
viability_diff %>% pivot_longer(cols = c(c, t)) %>% filter(age == "young") %>%
ggplot(aes(x = name, y = value)) +
geom_line(aes(group = male_ID, colour = mito)) +
scale_colour_manual(values = met3) +
facet_grid(mito ~ nuclear) +
NULL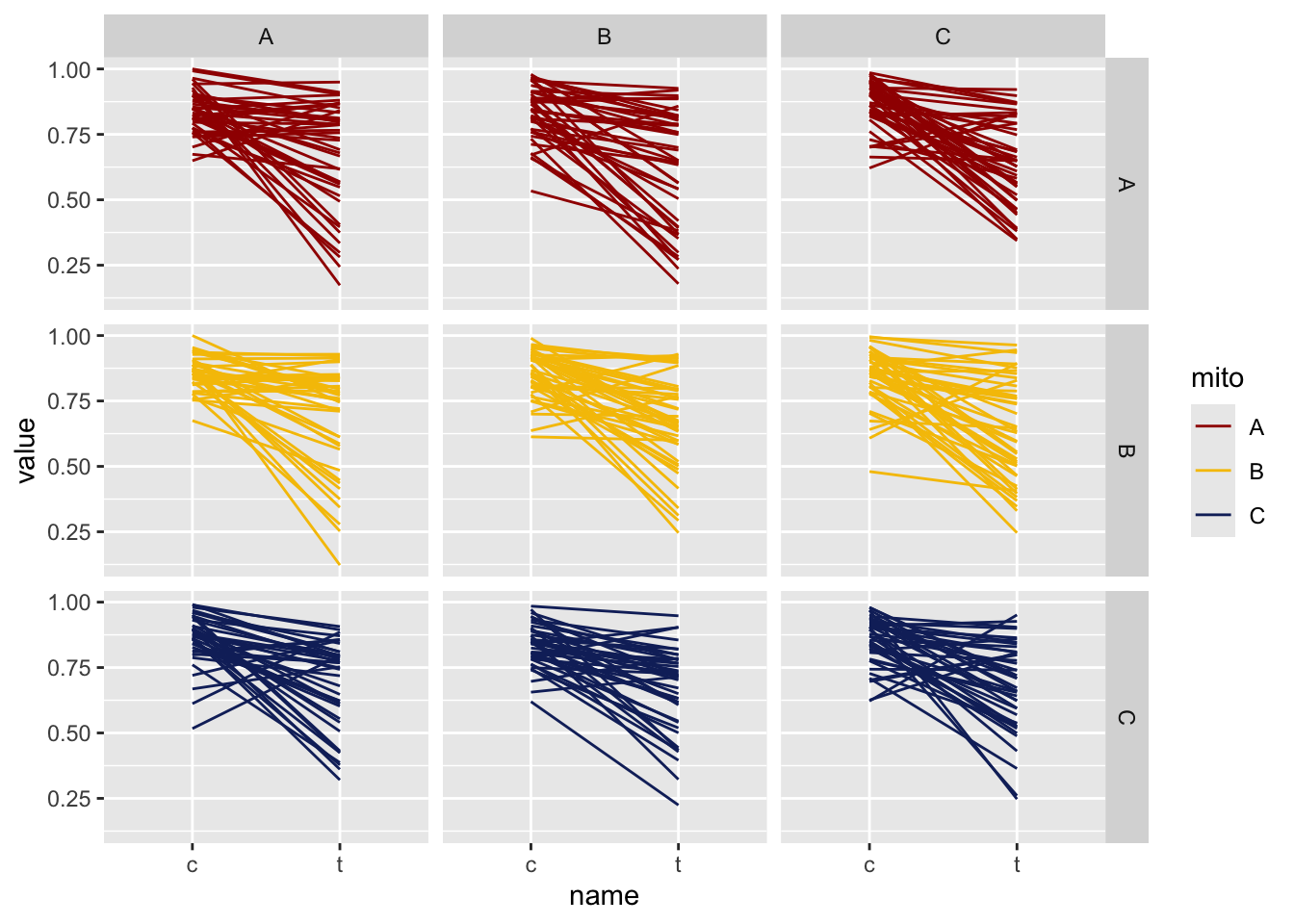
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
>>> Old males
viability_diff %>% pivot_longer(cols = c(c, t)) %>% filter(age == "old") %>%
ggplot(aes(x = name, y = value)) +
geom_line(aes(group = male_ID, colour = mito)) +
scale_colour_manual(values = met3) +
facet_grid(mito ~ nuclear) +
NULL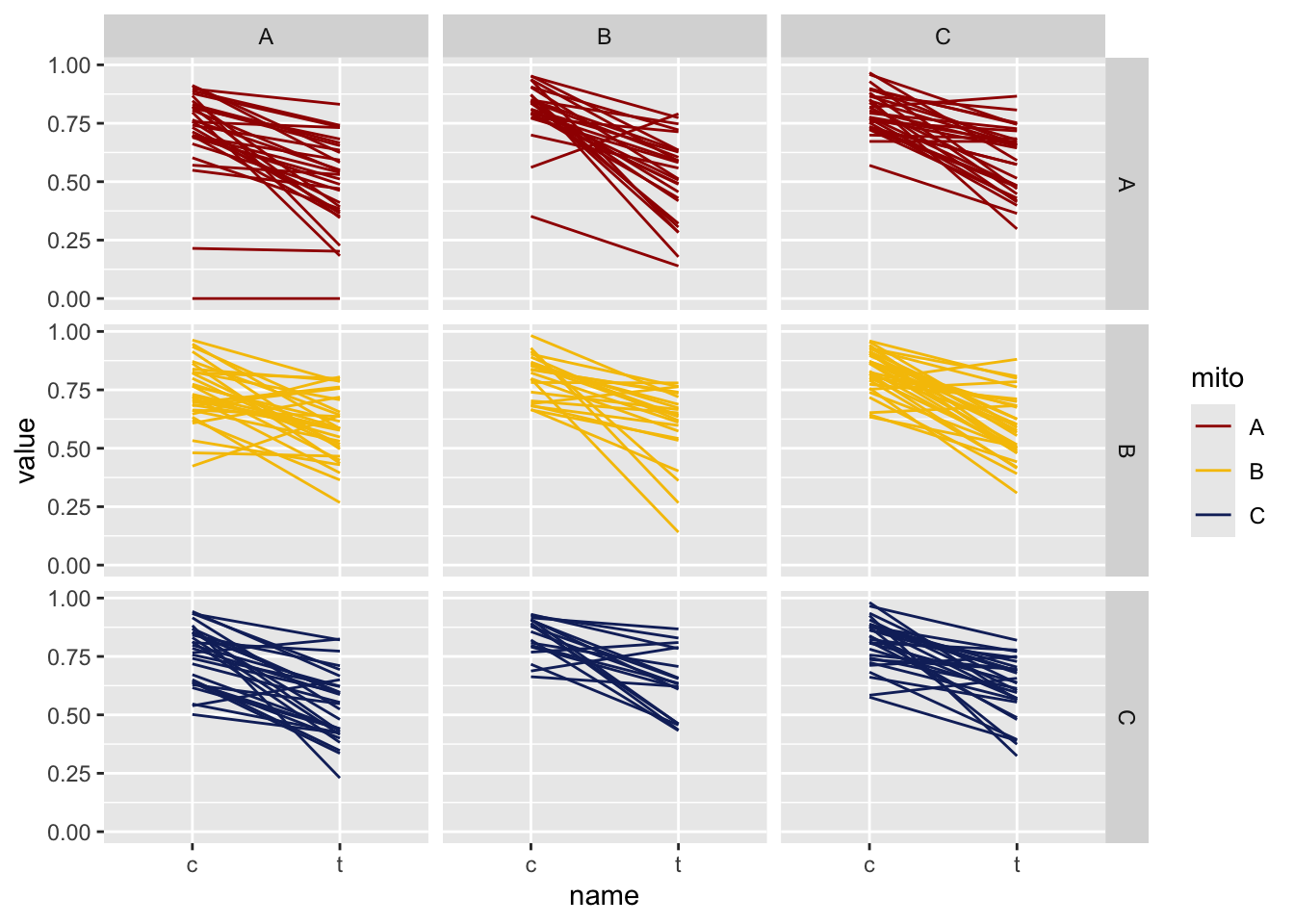
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
hist(viability_diff$diff)
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
vib_diff_test <- lmerTest::lmer(diff ~ mito * nuclear * age + block + days + (1 + age|LINE),
data = viability_diff)>>> Check diagnostics
performance::check_model(vib_diff_test)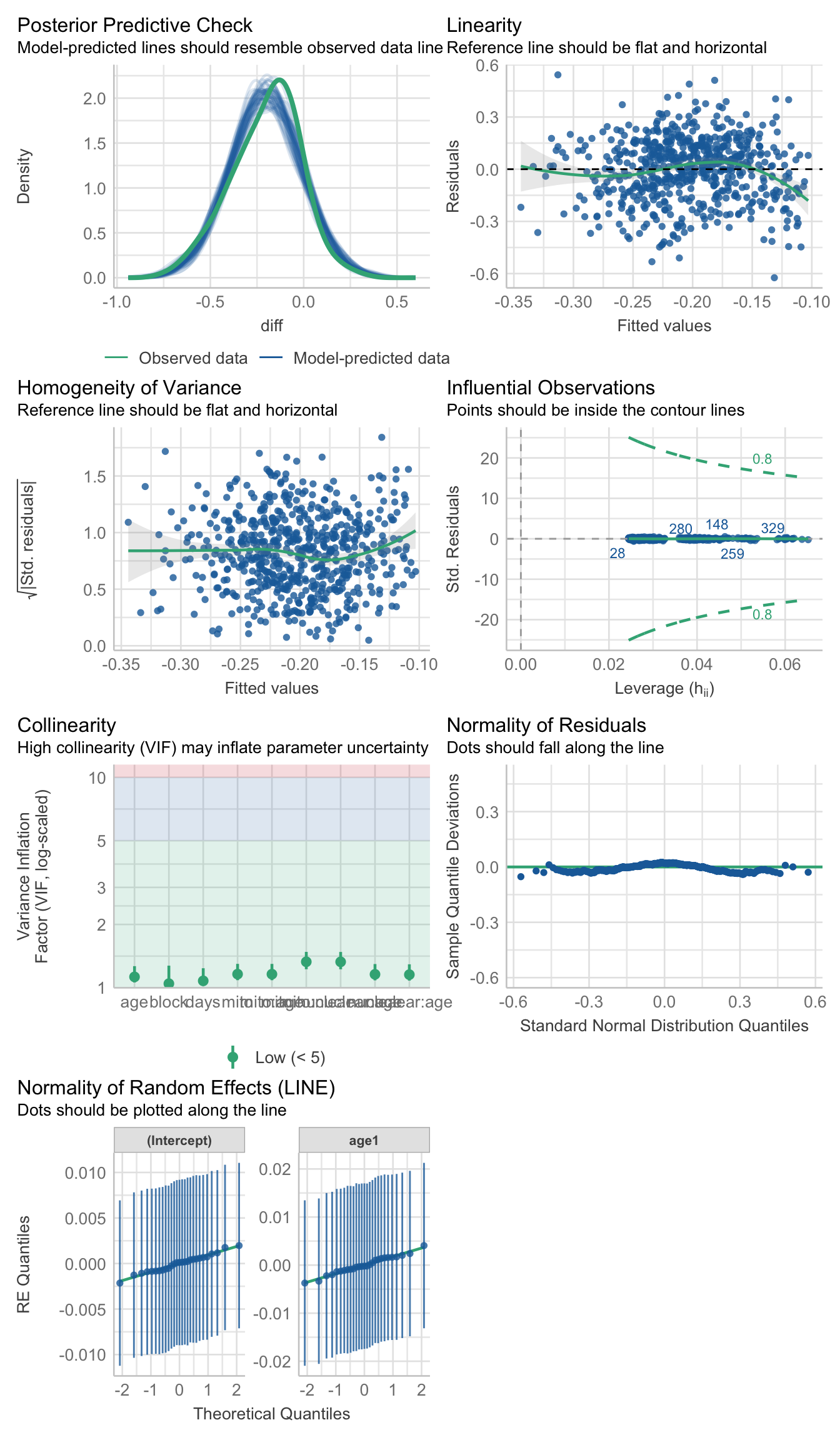
testDispersion(vib_diff_test)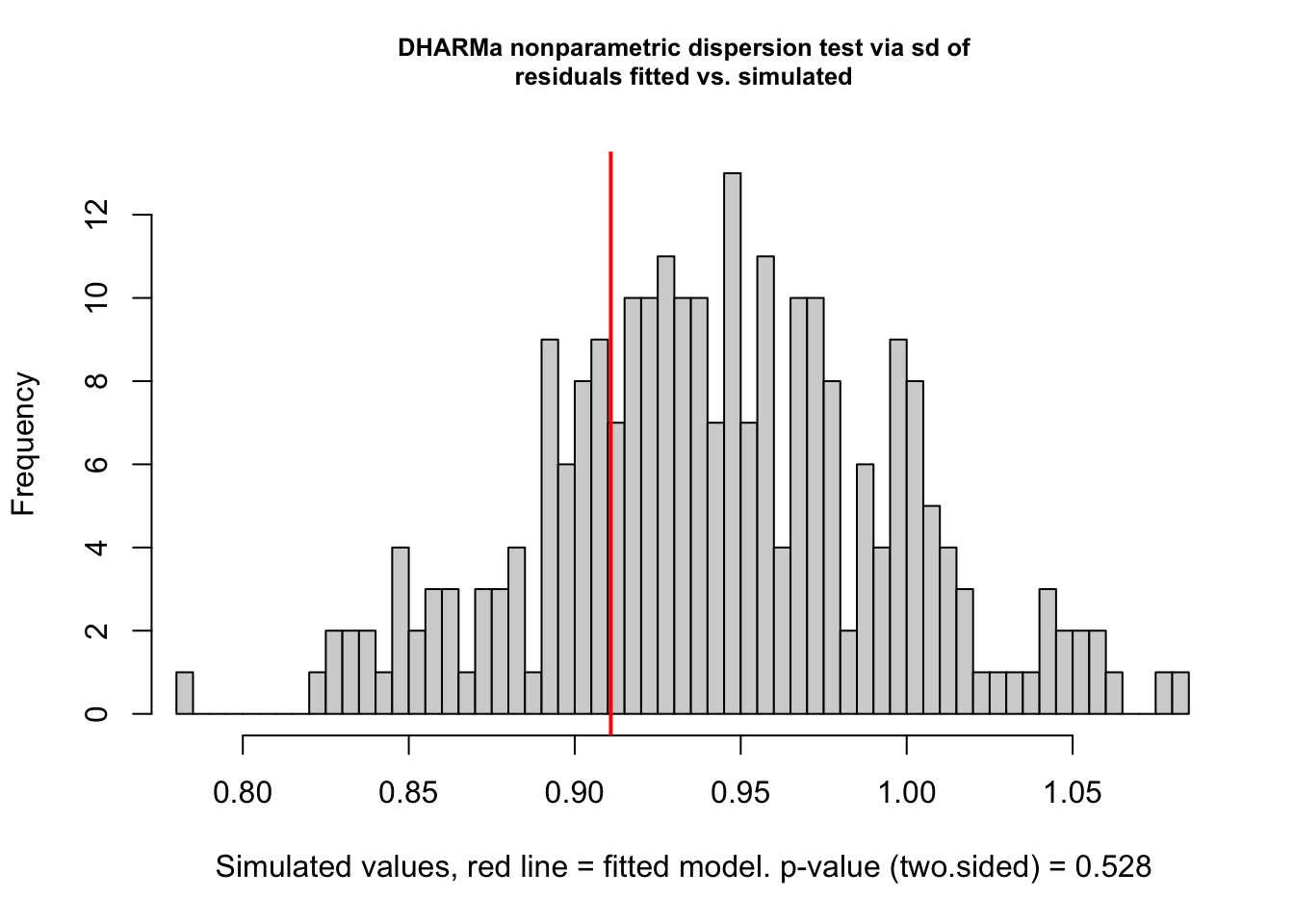
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
DHARMa nonparametric dispersion test via sd of residuals fitted vs.
simulated
data: simulationOutput
dispersion = 0.96613, p-value = 0.528
alternative hypothesis: two.sided
simulationOutput <- simulateResiduals(fittedModel = vib_diff_test, plot = FALSE)
hist(residuals(simulationOutput))
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
hist(residuals(simulationOutput, quantileFunction = qnorm, outlierValues = c(-7,7)))
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
plot(simulationOutput)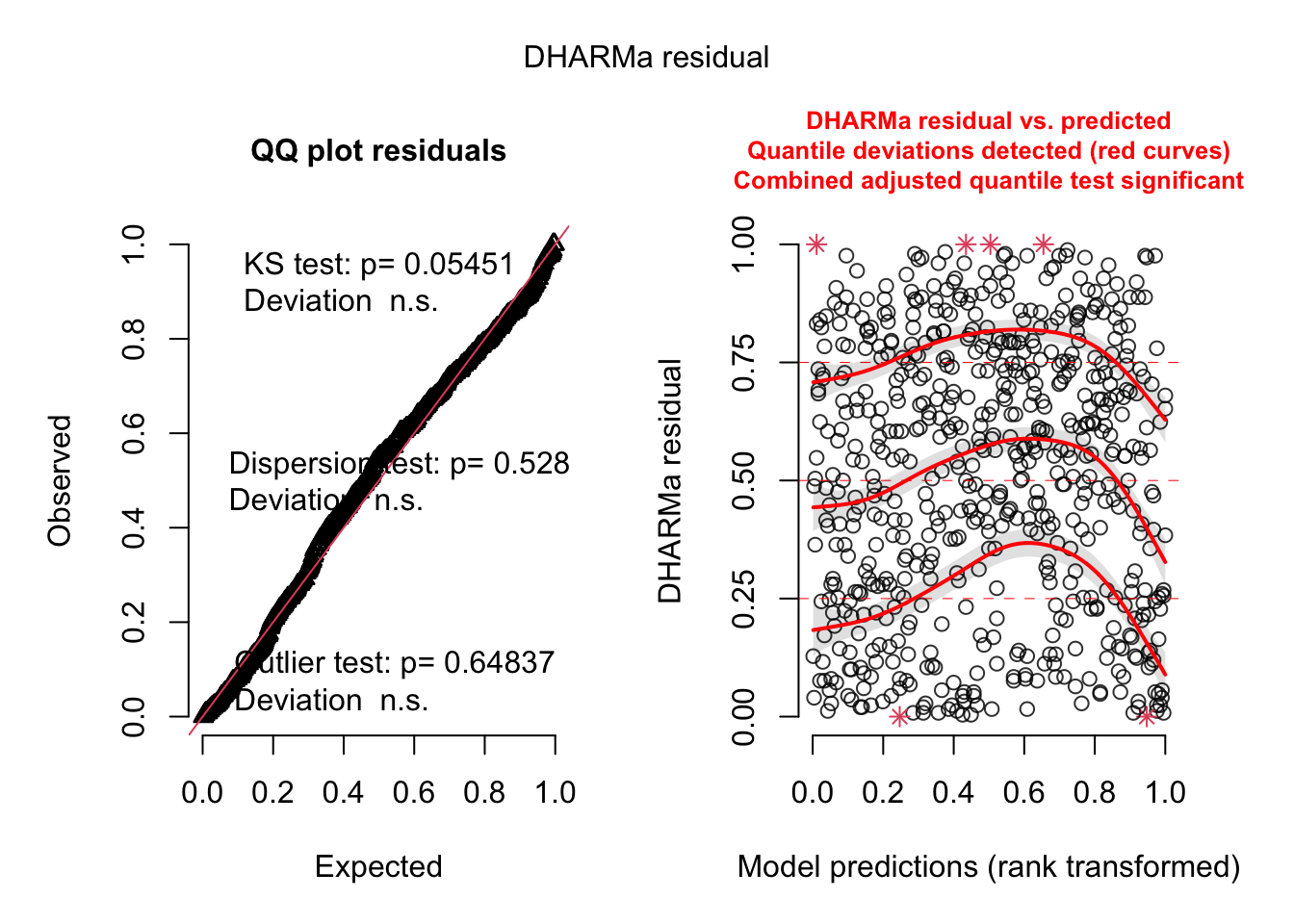
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
>>> Results
anova(vib_diff_test, type = "III", ddf = "Kenward-Roger") %>%
broom::tidy() %>%
as_tibble() %>%
#write_csv("output/anova_tables/sperm_quality.csv") %>% # save anova table for supp. tables
kable(digits = 3,
caption = 'Type III Analysis of Variance Table with Kenward-Roger`s method') %>%
kable_styling(full_width = FALSE)| term | sumsq | meansq | NumDF | DenDF | statistic | p.value |
|---|---|---|---|---|---|---|
| mito | 0.128 | 0.064 | 2 | 18.585 | 1.894 | 0.178 |
| nuclear | 0.130 | 0.065 | 2 | 18.471 | 1.915 | 0.176 |
| age | 0.315 | 0.315 | 1 | 22.638 | 9.312 | 0.006 |
| block | 0.345 | 0.345 | 1 | 579.154 | 10.204 | 0.001 |
| days | 0.255 | 0.255 | 1 | 576.522 | 7.549 | 0.006 |
| mito:nuclear | 0.172 | 0.043 | 4 | 18.333 | 1.271 | 0.317 |
| mito:age | 0.025 | 0.013 | 2 | 18.551 | 0.376 | 0.692 |
| nuclear:age | 0.030 | 0.015 | 2 | 18.432 | 0.447 | 0.646 |
| mito:nuclear:age | 0.093 | 0.023 | 4 | 18.331 | 0.686 | 0.611 |
#summary(vib_diff_test)
# posthoc tests
bind_rows(emmeans(vib_diff_test, pairwise ~ age, adjust = "tukey")$contrasts %>% broom::tidy(),
emmeans(vib_diff_test, pairwise ~ block, adjust = "tukey")$contrasts %>% broom::tidy()) %>%
kable(digits = 3,
caption = 'Posthoc Tukey tests to compare which groups differ. Results are averaged over the levels of: mito, nuclear, age, block') %>%
kable_styling(full_width = FALSE) %>%
kableExtra::group_rows("Age", 1, 1) %>%
kableExtra::group_rows("Block", 2, 2)| term | contrast | null.value | estimate | std.error | df | statistic | p.value |
|---|---|---|---|---|---|---|---|
| Age | |||||||
| age | young - old | 0 | 0.050 | 0.016 | 22.638 | 3.051 | 0.006 |
| Block | |||||||
| block | block1 - block2 | 0 | 0.048 | 0.015 | 579.154 | 3.194 | 0.001 |
emtrends(vib_diff_test, ~ days, var = "days") %>% broom::tidy() %>%
kable(digits = 3,
caption = 'Posthoc Tukey tests for days effect. Results are averaged over the levels of: mito, nuclear, age, block') %>%
kable_styling(full_width = FALSE)| days | days.trend | std.error | df | statistic | p.value |
|---|---|---|---|---|---|
| 3.738 | -0.01 | 0.004 | 576.522 | -2.747 | 0.006 |
>>>> Reaction norms
sperm_qual_norm <- emmeans(vib_diff_test, ~ mito * nuclear * age) %>% as_tibble() %>%
ggplot(aes(x = nuclear, y = emmean, fill = mito)) +
geom_line(aes(group = mito, colour = mito), position = position_dodge(width = .5)) +
# geom_errorbar(aes(ymin = lower.CL, ymax = upper.CL),
# width = .25, position = position_dodge(width = .5)) +
geom_point(size = 3, pch = 21, position = position_dodge(width = .5)) +
labs(y = 'Sperm viability (EMM)') +
scale_colour_manual(values = met3) +
scale_fill_manual(values = met3) +
facet_wrap(~ age) +
theme_bw() +
theme() +
NULL
sperm_qual_norm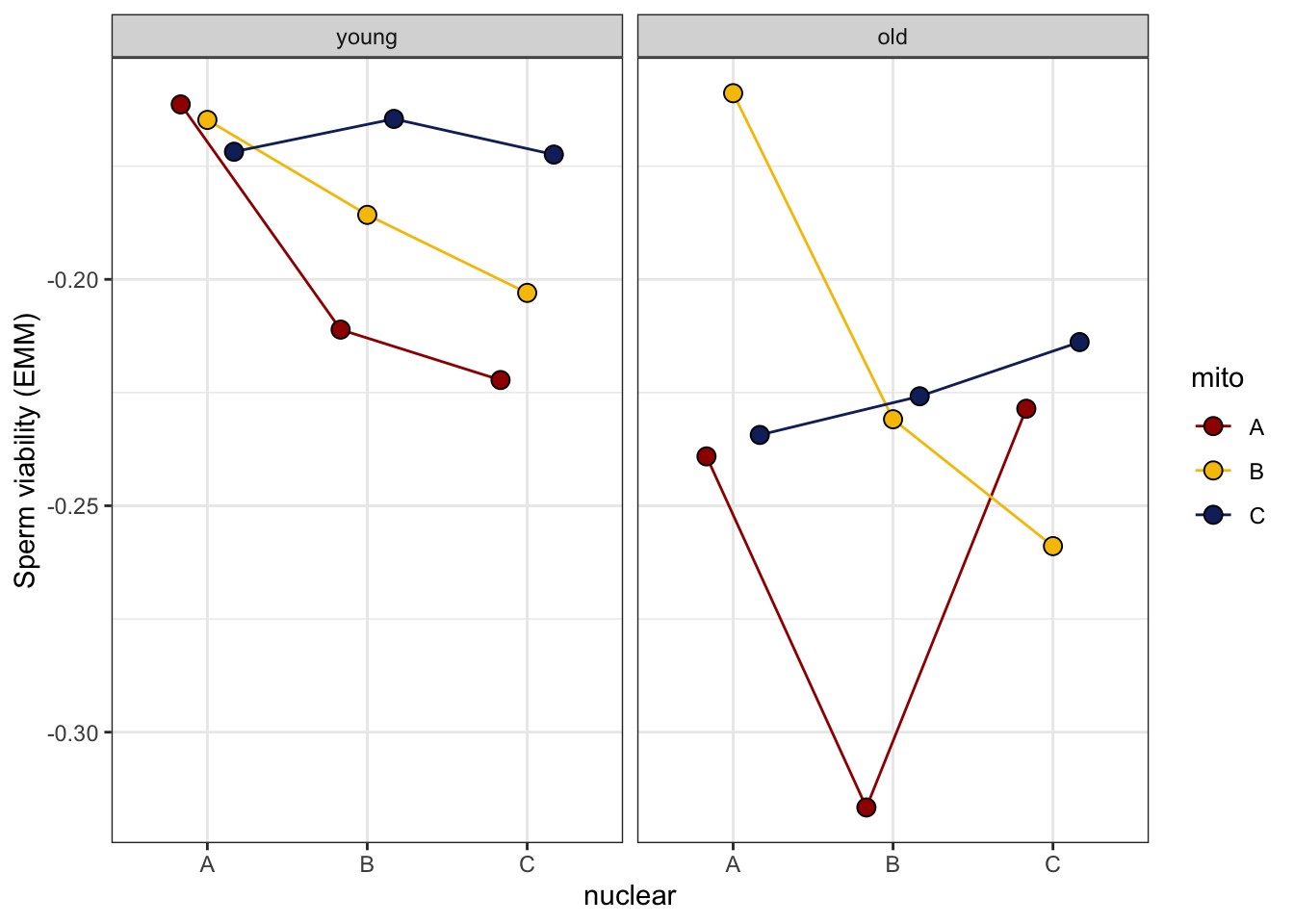
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
>>> Matched vs. mismatched
vib_diff_coevo <- lmerTest::lmer(diff ~ coevolved * age + block + days + (age|LINE),
data = viability_diff,
na.action = 'na.fail')
anova(vib_diff_coevo, type = "III", ddf = "Kenward-Roger")Type III Analysis of Variance Table with Kenward-Roger's method
Sum Sq Mean Sq NumDF DenDF F value Pr(>F)
coevolved 0.01983 0.01983 1 24.82 0.5866 0.450964
age 0.27915 0.27915 1 29.18 8.2593 0.007489 **
block 0.33197 0.33197 1 586.41 9.8219 0.001811 **
days 0.25758 0.25758 1 579.82 7.6210 0.005952 **
coevolved:age 0.00253 0.00253 1 24.85 0.0748 0.786719
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
>>> Mito-type analysis
vib_diff_snp <- lmerTest::lmer(diff ~ mito_snp * nuclear * age + block + days + (1 + age|LINE),
data = viability_diff)
anova(vib_diff_snp, type = "III", ddf = "Kenward-Roger")Type III Analysis of Variance Table with Kenward-Roger's method
Sum Sq Mean Sq NumDF DenDF F value Pr(>F)
mito_snp 0.29901 0.03738 8 9.09 1.0899 0.445593
nuclear 0.07678 0.03839 2 8.96 1.1188 0.368350
age 0.38706 0.38706 1 10.43 11.2829 0.006848 **
block 0.34418 0.34418 1 575.40 10.0328 0.001619 **
days 0.25922 0.25922 1 575.16 7.5564 0.006168 **
mito_snp:nuclear 0.10879 0.01554 7 9.08 0.4530 0.845633
mito_snp:age 0.23540 0.02942 8 9.09 0.8580 0.579924
nuclear:age 0.01427 0.00714 2 8.95 0.2079 0.816073
mito_snp:nuclear:age 0.14458 0.02065 7 9.08 0.6020 0.742017
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
> Sperm metabolic rate
We measured the metabolic rate of sperm dissected from seminal vesicles using NAD(P)H autofluorescence lifetime imaging (FLIM). Specifically, here we use parameter a2 (see online supplementary material for detailed methods).
Looking at the data there is a clear outlier in old males where
a2 > 50. We will analyse the data including and
excluding this outlier.
sperm_met %>%
group_by(mito, nuclear, age) %>%
summarise(mn = mean(a2),
se = sd(a2)/sqrt(n()),
s95 = se * 1.96) %>%
ggplot(aes(x = nuclear, y = mn, fill = mito)) +
geom_point(data = sperm_met,
aes(y = a2, colour = mito), alpha = .25,
position = position_jitterdodge(dodge.width = .5, jitter.width = .1)) +
geom_errorbar(aes(ymin = mn - s95, ymax = mn + s95),
width = .25, position = position_dodge(width = .5)) +
geom_point(size = 3, pch = 21, position = position_dodge(width = .5)) +
labs(y = 'a2') +
facet_wrap(~ age, scales = 'free_x') +
scale_colour_manual(values = met3) +
scale_fill_manual(values = met3) +
theme_bw() +
theme() +
NULL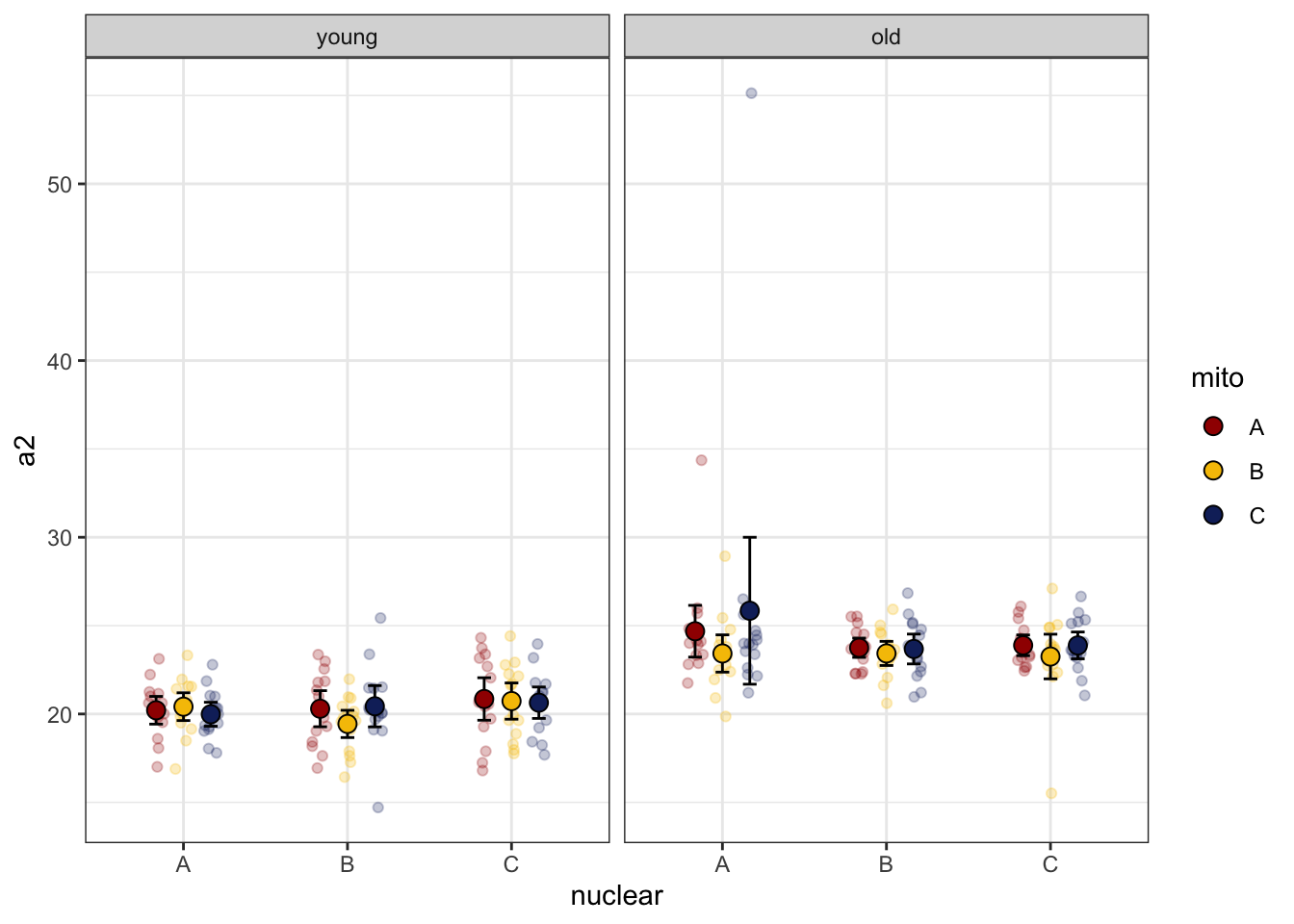
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
>> Analysis
hist(sperm_met$p.a2, breaks = 50)
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
>>> With outlier
met_mod_outlier_test <- lmerTest::lmer(p.a2 ~ mito * nuclear * age + (1 + age|LINE),
data = sperm_met, REML = TRUE)
anova(met_mod_outlier_test, type = "III", ddf = "Kenward-Roger")Type III Analysis of Variance Table with Kenward-Roger's method
Sum Sq Mean Sq NumDF DenDF F value Pr(>F)
mito 0.001762 0.000881 2 17.996 1.2674 0.3055
nuclear 0.001419 0.000710 2 17.996 1.0206 0.3803
age 0.086787 0.086787 1 17.996 124.8329 1.584e-09 ***
mito:nuclear 0.000415 0.000104 4 17.995 0.1492 0.9609
mito:age 0.000956 0.000478 2 17.995 0.6876 0.5155
nuclear:age 0.002506 0.001253 2 17.995 1.8020 0.1935
mito:nuclear:age 0.002388 0.000597 4 17.994 0.8586 0.5072
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
>>> Without outlier
met_mod_test <- lmerTest::lmer(p.a2 ~ mito * nuclear * age + (1 + age|LINE),
data = sperm_met %>% filter(a2 <= 30), REML = TRUE)>>>> Check diagnostics
performance::check_model(met_mod_outlier_test)
testDispersion(met_mod_outlier_test)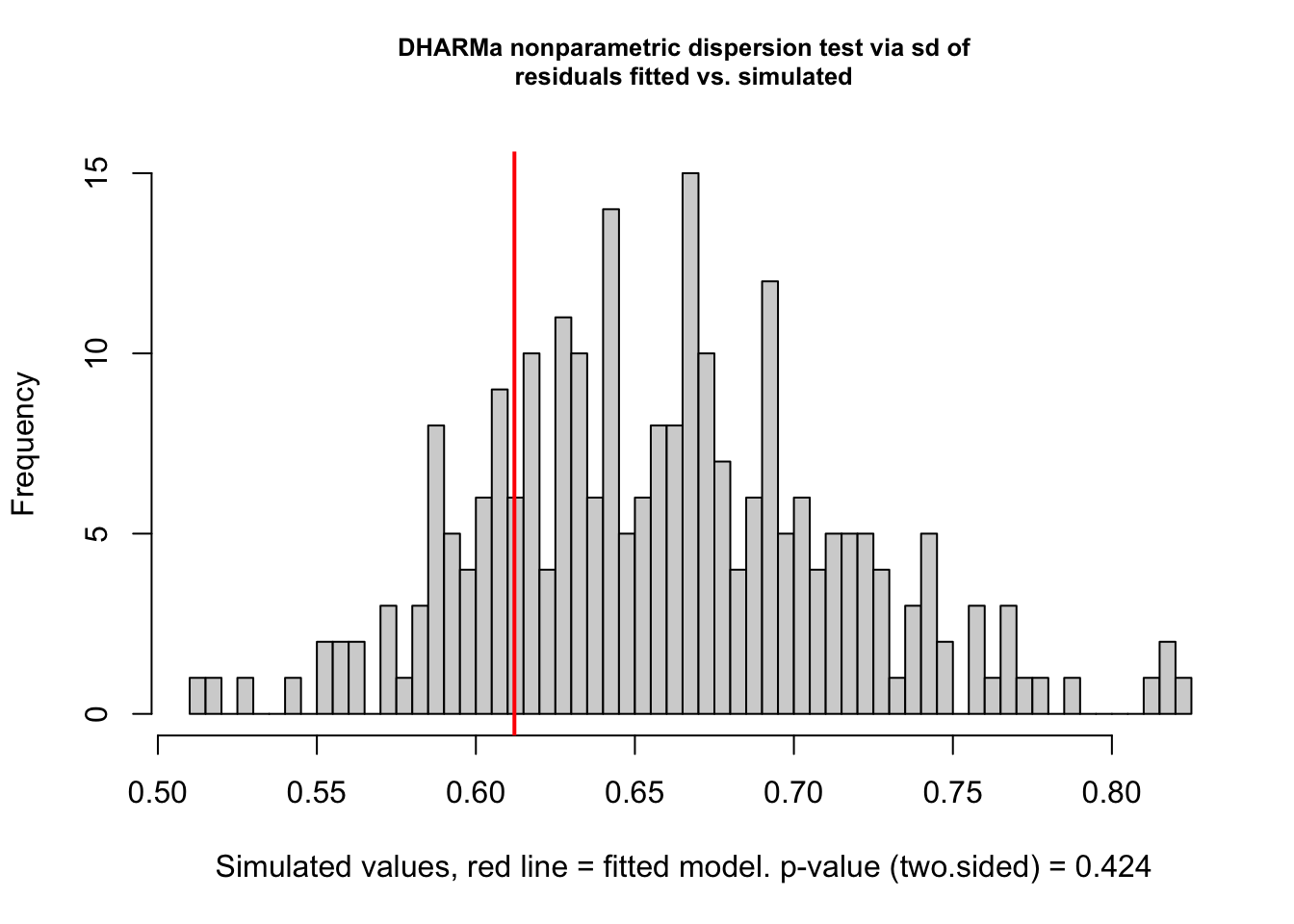
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
DHARMa nonparametric dispersion test via sd of residuals fitted vs.
simulated
data: simulationOutput
dispersion = 0.92929, p-value = 0.424
alternative hypothesis: two.sided
simulationOutput <- simulateResiduals(fittedModel = met_mod_outlier_test, plot = FALSE)
hist(residuals(simulationOutput))
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
hist(residuals(simulationOutput, quantileFunction = qnorm, outlierValues = c(-7,7)))
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
plot(simulationOutput)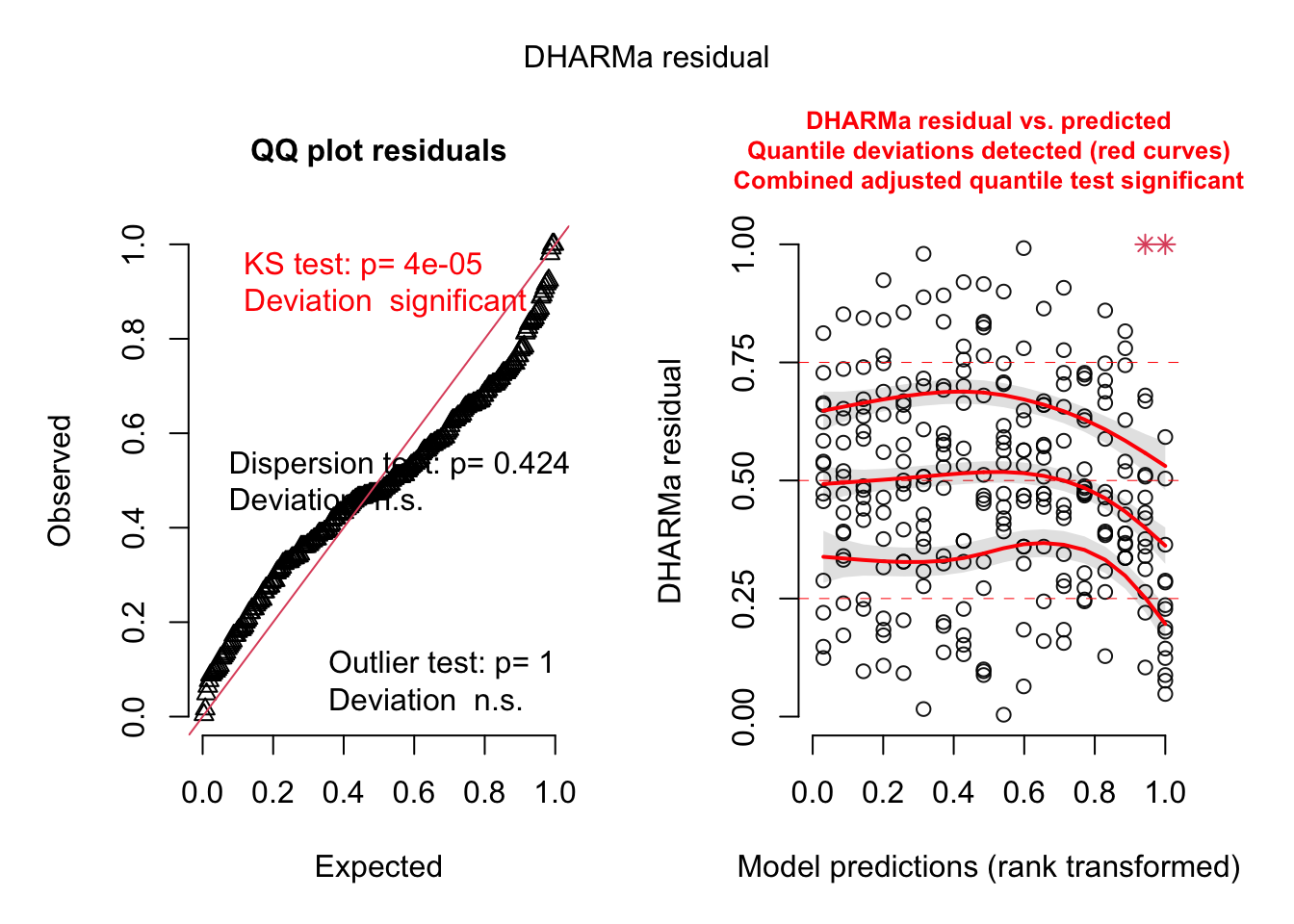
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
>> Results
anova(met_mod_test, type = "III", ddf = "Kenward-Roger") %>%
broom::tidy() %>%
as_tibble() #%>% write_csv("output/anova_tables/sperm_metabolism.csv") %>% # save anova table for supp. tables# A tibble: 7 × 7 term sumsq meansq NumDF DenDF statistic p.value1 mito 0.000699 0.000350 2 18.0 1.16 3.37e- 1 2 nuclear 0.000625 0.000313 2 18.0 1.04 3.75e- 1 3 age 0.0726 0.0726 1 18.0 240. 7.51e-12 4 mito:nuclear 0.000341 0.0000852 4 18.0 0.282 8.86e- 1 5 mito:age 0.0000876 0.0000438 2 18.0 0.145 8.66e- 1 6 nuclear:age 0.000535 0.000268 2 18.0 0.886 4.29e- 1 7 mito:nuclear:age 0.000611 0.000153 4 18.0 0.506 7.32e- 1
met_emm <- emmeans(met_mod_test, ~ mito * nuclear * age)
emmeans(met_mod_test, pairwise ~ age, adjust = "tukey")$emmeans age emmean SE df lower.CL upper.CL young 0.203 0.00153 18.0 0.200 0.207 old 0.237 0.00150 17.9 0.234 0.240 Results are averaged over the levels of: mito, nuclear Degrees-of-freedom method: kenward-roger Confidence level used: 0.95 $contrasts contrast estimate SE df t.ratio p.value young - old -0.0334 0.00215 18 -15.503 <.0001 Results are averaged over the levels of: mito, nuclear Degrees-of-freedom method: kenward-roger
>>> Reaction norms
sperm_met_norm <- emmeans(met_mod_test, ~ mito * nuclear * age) %>% as_tibble() %>%
ggplot(aes(x = nuclear, y = emmean, fill = mito)) +
geom_line(aes(group = mito, colour = mito), position = position_dodge(width = .5)) +
# geom_errorbar(aes(ymin = lower.CL, ymax = upper.CL),
# width = .25, position = position_dodge(width = .5)) +
geom_point(size = 3, pch = 21, position = position_dodge(width = .5)) +
labs(y = 'Sperm metabolic rate, a2 (EMM)') +
scale_colour_manual(values = met3) +
scale_fill_manual(values = met3) +
facet_wrap(~ age) +
theme_bw() +
theme() +
NULL
sperm_met_norm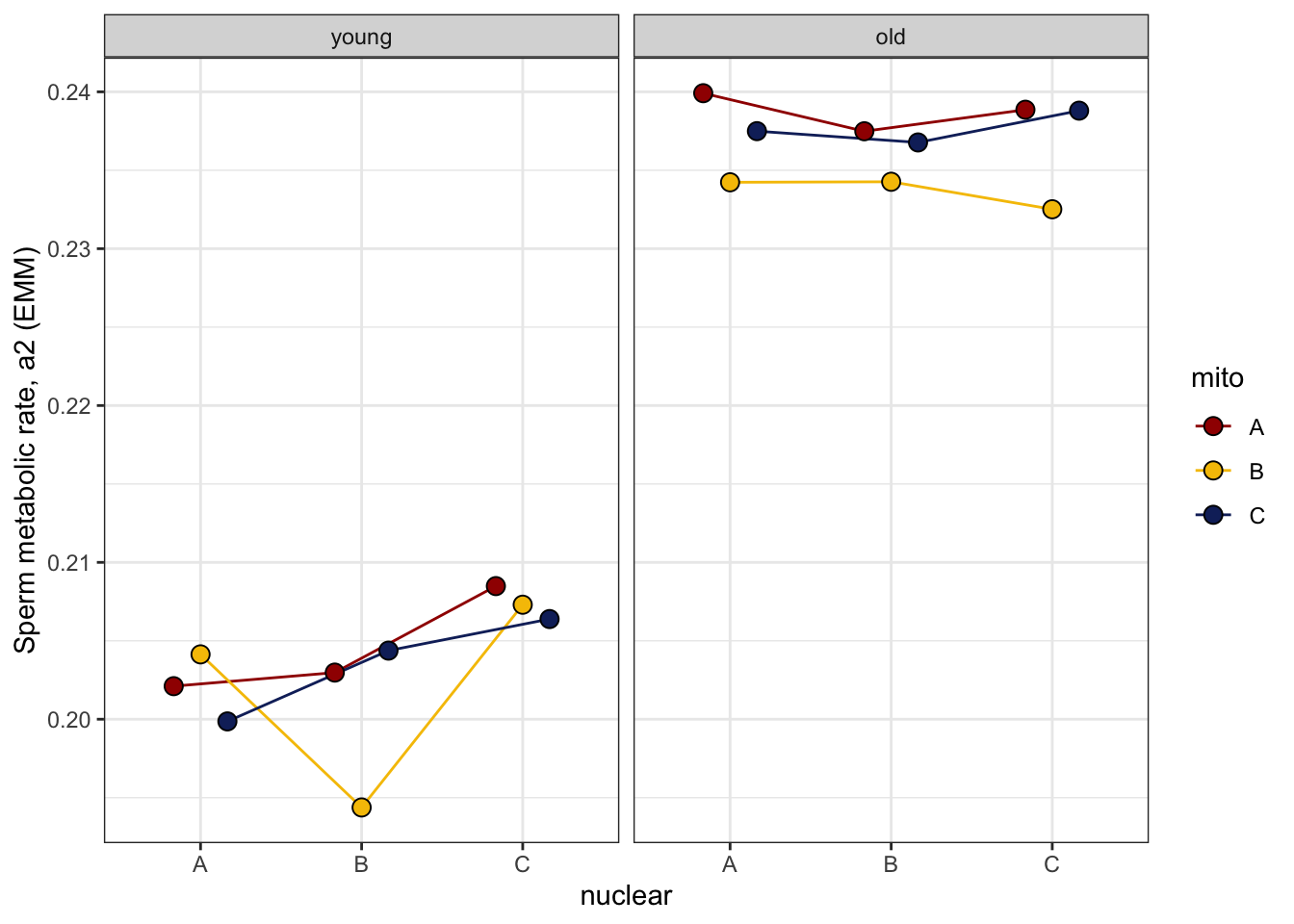
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
>>> Raw data and means
sperm_met_raw <- emmeans(met_mod_test, ~ mito * nuclear * age) %>% as_tibble() %>%
ggplot(aes(x = nuclear, y = emmean, fill = mito)) +
geom_jitter(data = sperm_met,
aes(y = p.a2, colour = mito),
position = position_jitterdodge(dodge.width = .5, jitter.width = .1),
size = 0.75, alpha = .1) +
geom_jitter(data = sperm_met %>%
group_by(LINE, age) %>%
summarise(mn = mean(p.a2)) %>%
separate(LINE, into = c("mito", "nuclear", NA), sep = "(?<=.)", remove = FALSE),
aes(y = mn, colour = mito),
position = position_jitterdodge(dodge.width = .5, jitter.width = .1),
alpha = .85, size = 2) +
geom_errorbar(aes(ymin = lower.CL, ymax = upper.CL),
width = .25, position = position_dodge(width = .5)) +
geom_point(size = 3, pch = 21, position = position_dodge(width = .5)) +
labs(y = 'Sperm metabolic rate, a2 (EMM ± 95% CIs)') +
scale_colour_manual(values = met3) +
scale_fill_manual(values = met3) +
coord_cartesian(ylim = c(0.14, 0.34)) +
facet_wrap(~ age) +
theme_bw() +
theme() +
ggrepel::geom_text_repel(data = sperm_met %>% filter(a2 > 50), aes(y = p.a2), label = "CA1, a2 = 0.55") +
geom_text(data = sperm_met %>% group_by(mito, nuclear, age) %>% count(), aes(y = 0.14, label = n),
size = 2, position = position_dodge(width = .5)) +
#ggsave('figures/sperm_met_raw.pdf', height = 4, width = 8, dpi = 600, useDingbats = FALSE) +
NULL
sperm_met_raw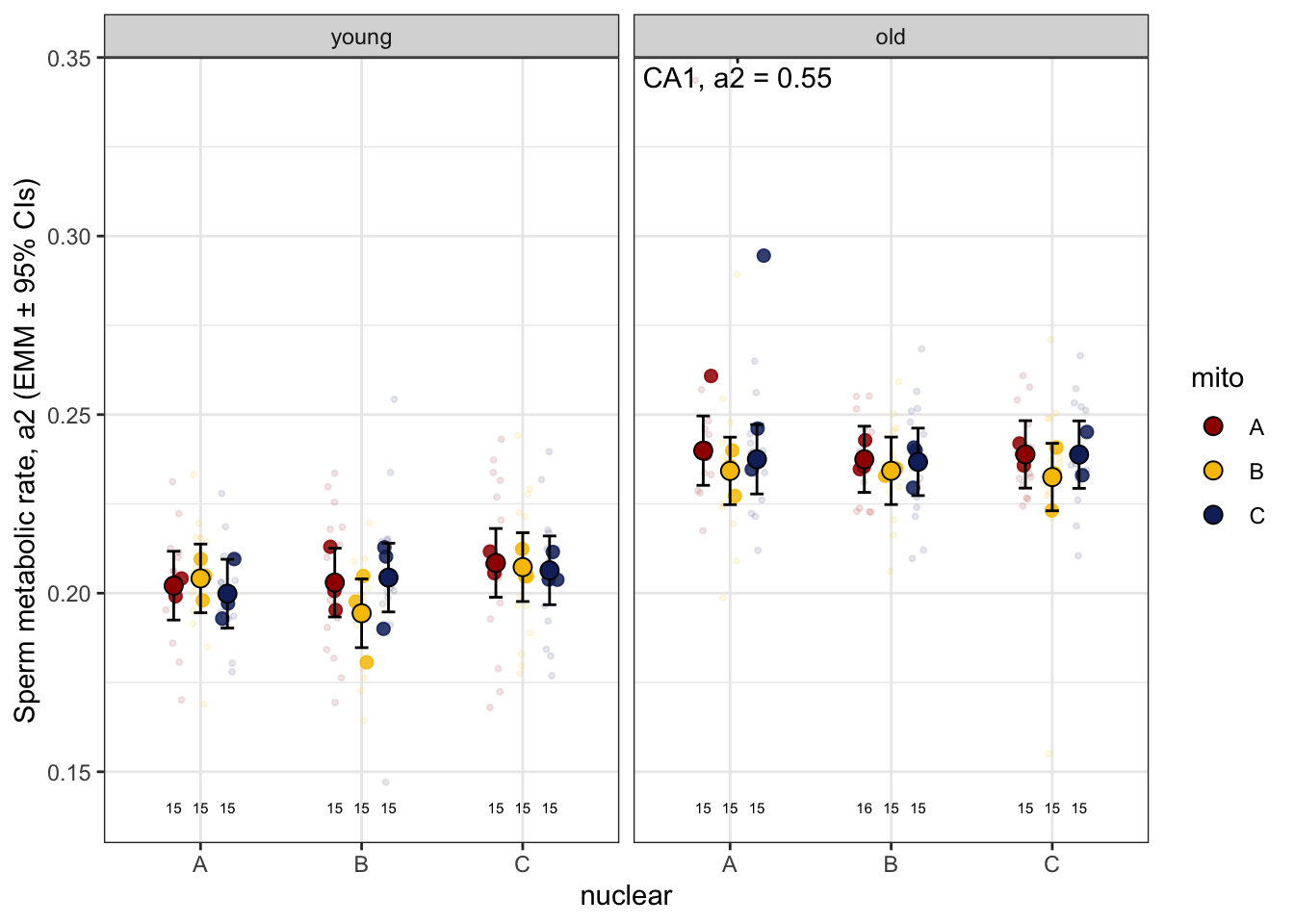
>>> Matched vs. mismatched
met_mod_coevo <- lmerTest::lmer(p.a2 ~ coevolved * age + (1 + age|LINE),
data = sperm_met %>% filter(a2 <= 30), REML = TRUE)
anova(met_mod_coevo, type = "III", ddf = "Kenward-Roger")Type III Analysis of Variance Table with Kenward-Roger's method
Sum Sq Mean Sq NumDF DenDF F value Pr(>F)
coevolved 0.000071 0.000071 1 25.163 0.2392 0.6290
age 0.069532 0.069532 1 25.163 234.5554 2.905e-14 ***
coevolved:age 0.000364 0.000364 1 25.163 1.2289 0.2781
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
>>> Mito-type analysis
met_mod_outlier_rm_snp <- lmerTest::lmer(p.a2 ~ mito_snp * nuclear * age + (1 + age|LINE),
data = sperm_met %>% filter(a2 <= 30))
anova(met_mod_outlier_rm_snp, type = "III", ddf = "Kenward-Roger")Type III Analysis of Variance Table with Kenward-Roger's method
Sum Sq Mean Sq NumDF DenDF F value Pr(>F)
mito_snp 0.001854 0.000232 8 8.8749 0.7690 0.6392
nuclear 0.001367 0.000683 2 8.8614 2.2672 0.1603
age 0.054751 0.054751 1 9.0178 181.6457 2.792e-07 ***
mito_snp:nuclear 0.002263 0.000323 7 8.8640 1.0724 0.4509
mito_snp:age 0.001489 0.000186 8 8.8749 0.6175 0.7456
nuclear:age 0.000634 0.000317 2 8.8614 1.0511 0.3894
mito_snp:nuclear:age 0.001933 0.000276 7 8.8640 0.9162 0.5358
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
> Sperm competition
We measured sperm defence (P1) and offence (P2) for focal males
against a competitor brown eyed (BW) male for 15 males from
each mitonuclear population. The data shows overdispersion, so we use
male ID as an observation level random effect. Note that
here, we only measured young (5 day old) males.
>> Sperm defence (P1)
First we take a look at the data for each Day and
Block, and see how P1 changes with copulation duration
(cop.dur).
>>> Day and block effect
defence %>%
ggplot(aes(x = nuclear, y = P1, colour = mito)) +
geom_jitter(position = position_jitterdodge(dodge.width = .65, jitter.width = .15),
alpha = .5) +
labs(y = 'Sperm viability (EMM ± 95% CIs)') +
scale_colour_manual(values = met3) +
scale_fill_manual(values = met3) +
theme_bw() +
theme() +
stat_summary(position = position_dodge(width = 0.65)) +
facet_grid(Day ~ Block) +
NULL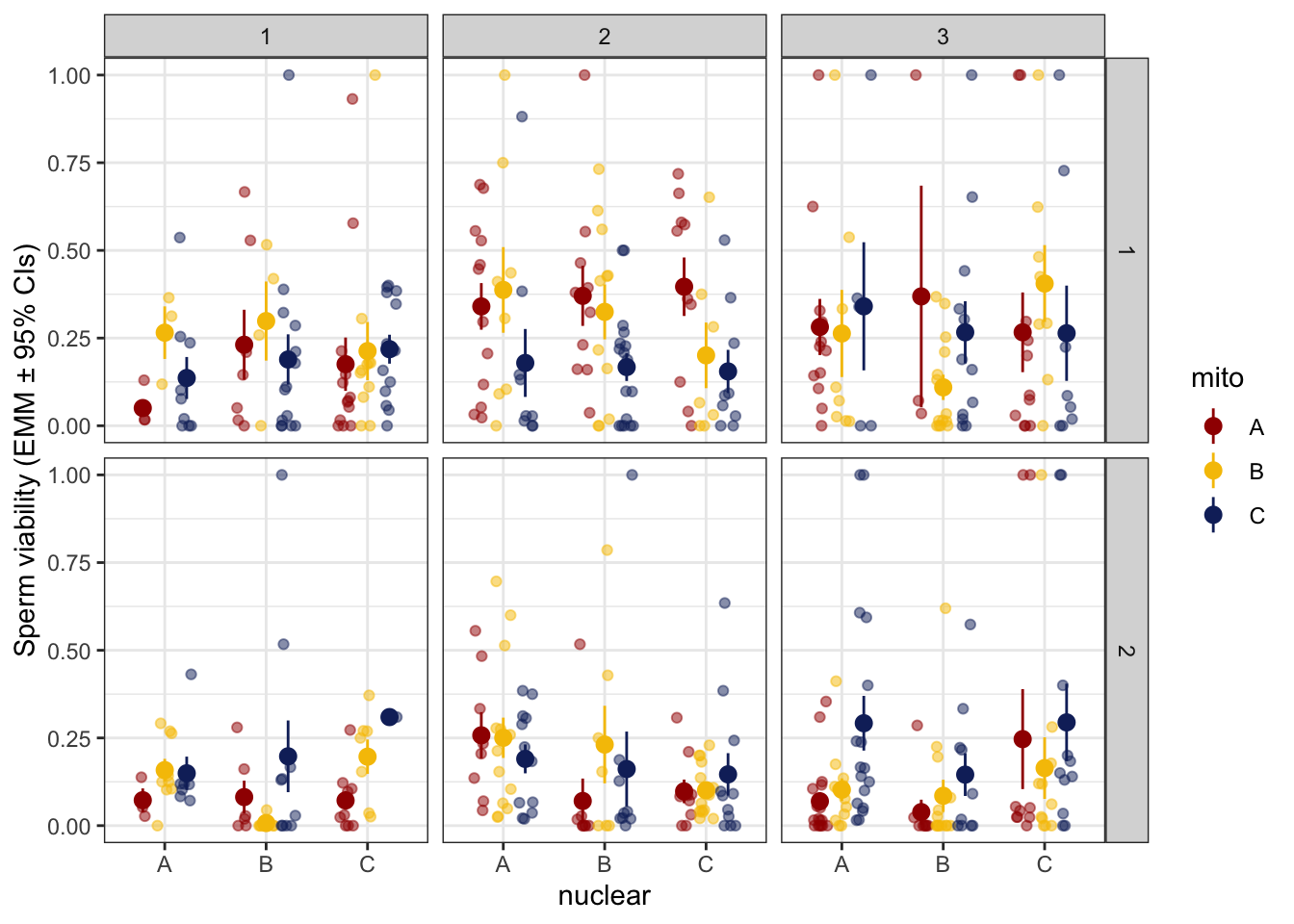
>>> Copulation duration
defence %>%
ggplot(aes(x = cop.dur, y = P1, colour = nuclear)) +
geom_smooth(method = "lm", se = FALSE) +
scale_colour_manual(values = met3) +
scale_fill_manual(values = met3) +
theme_bw() +
theme() +
facet_grid(Day ~ Block) +
NULL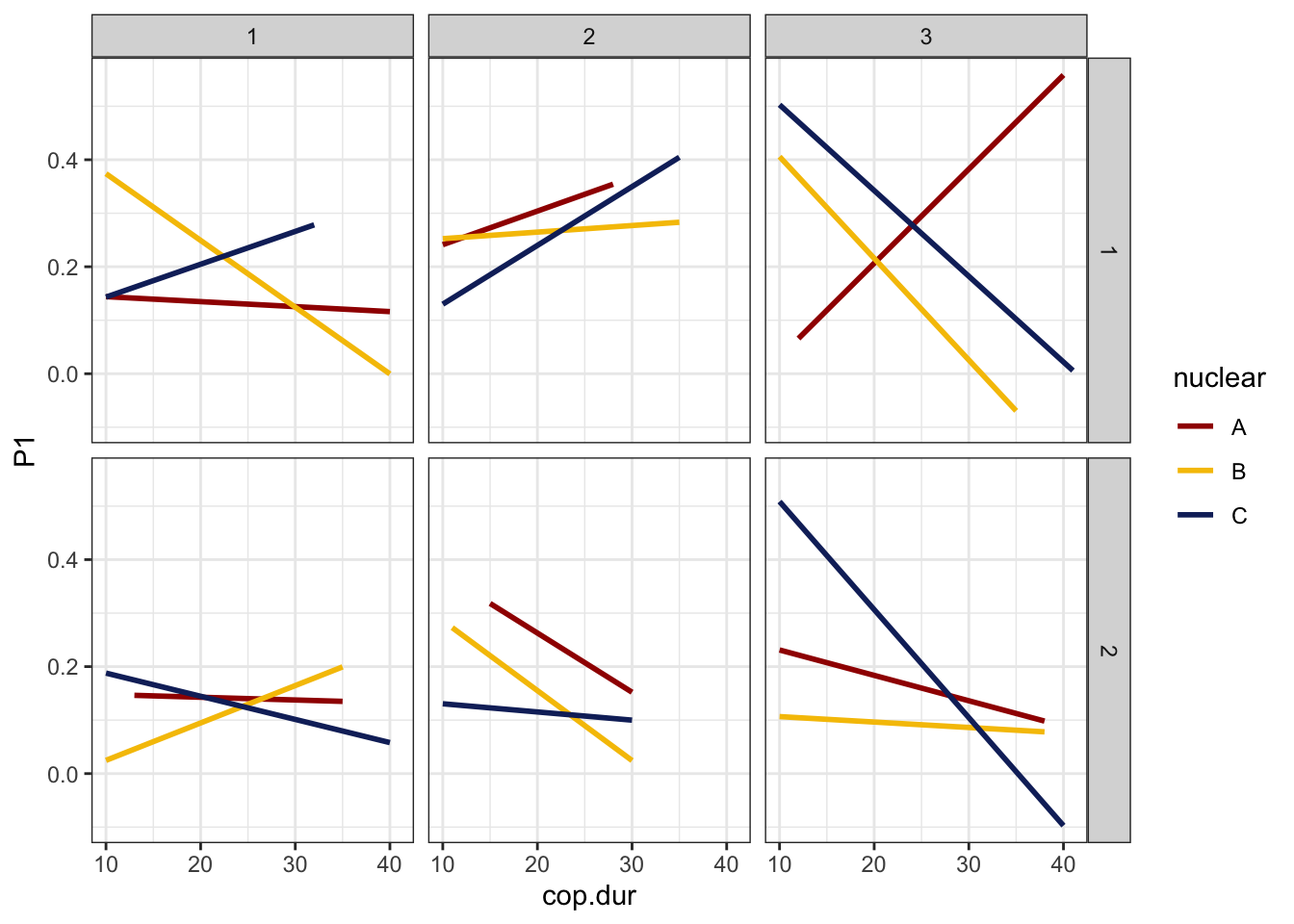
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
p1.mod1 <- glmer(cbind(RED, BW) ~ mito * nuclear + Day + cop.dur + (1 + cop.dur|LINE) + (1|ID) + (1|Block),
data = defence, family = binomial,
control= glmerControl(optimizer="bobyqa", optCtrl=list(maxfun=50000))
)>>> Check diagnostics
performance::check_model(p1.mod1)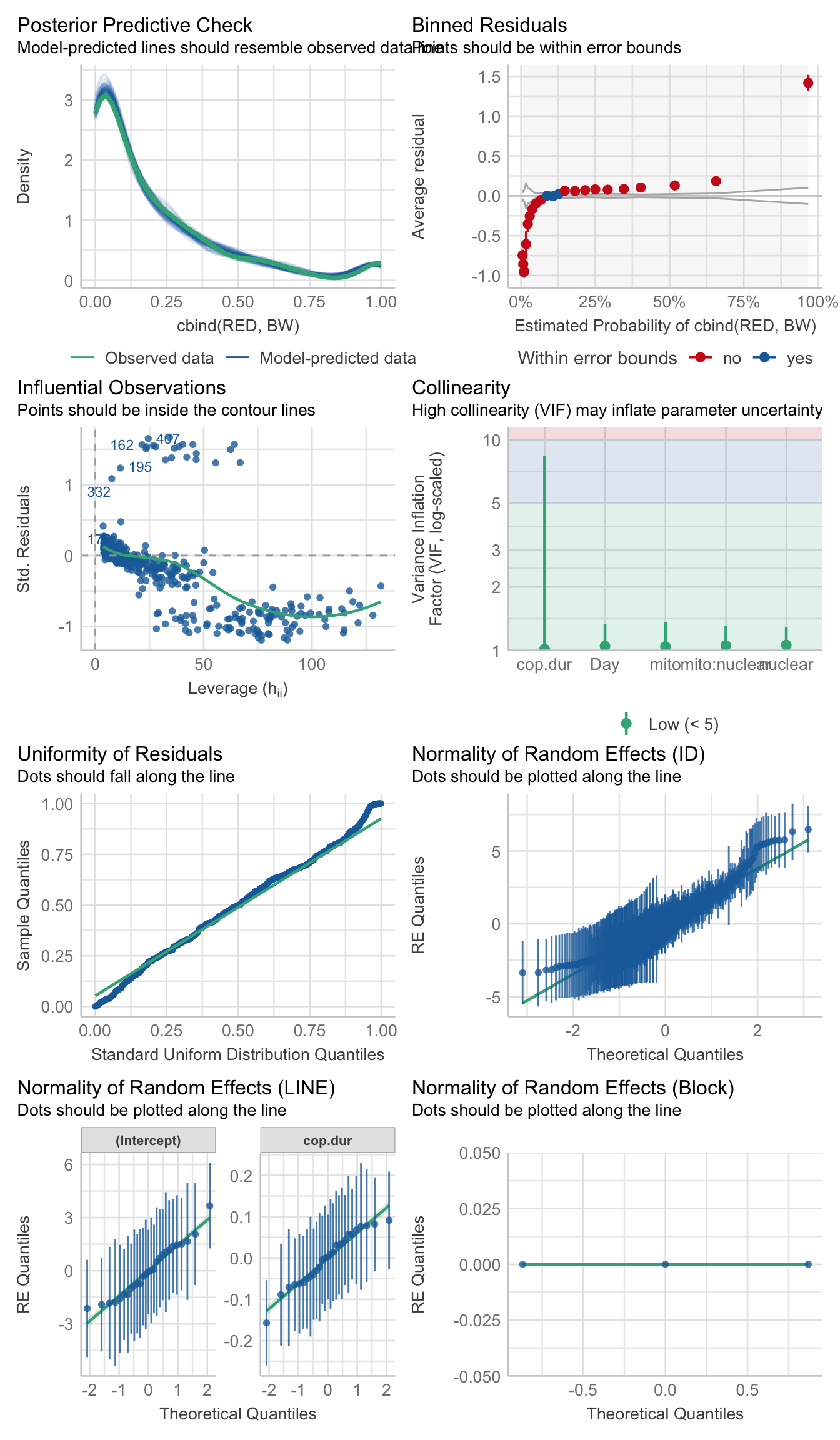
performance::check_overdispersion(p1.mod1)# Overdispersion test
dispersion ratio = 0.887
p-value = 0.224
testDispersion(p1.mod1)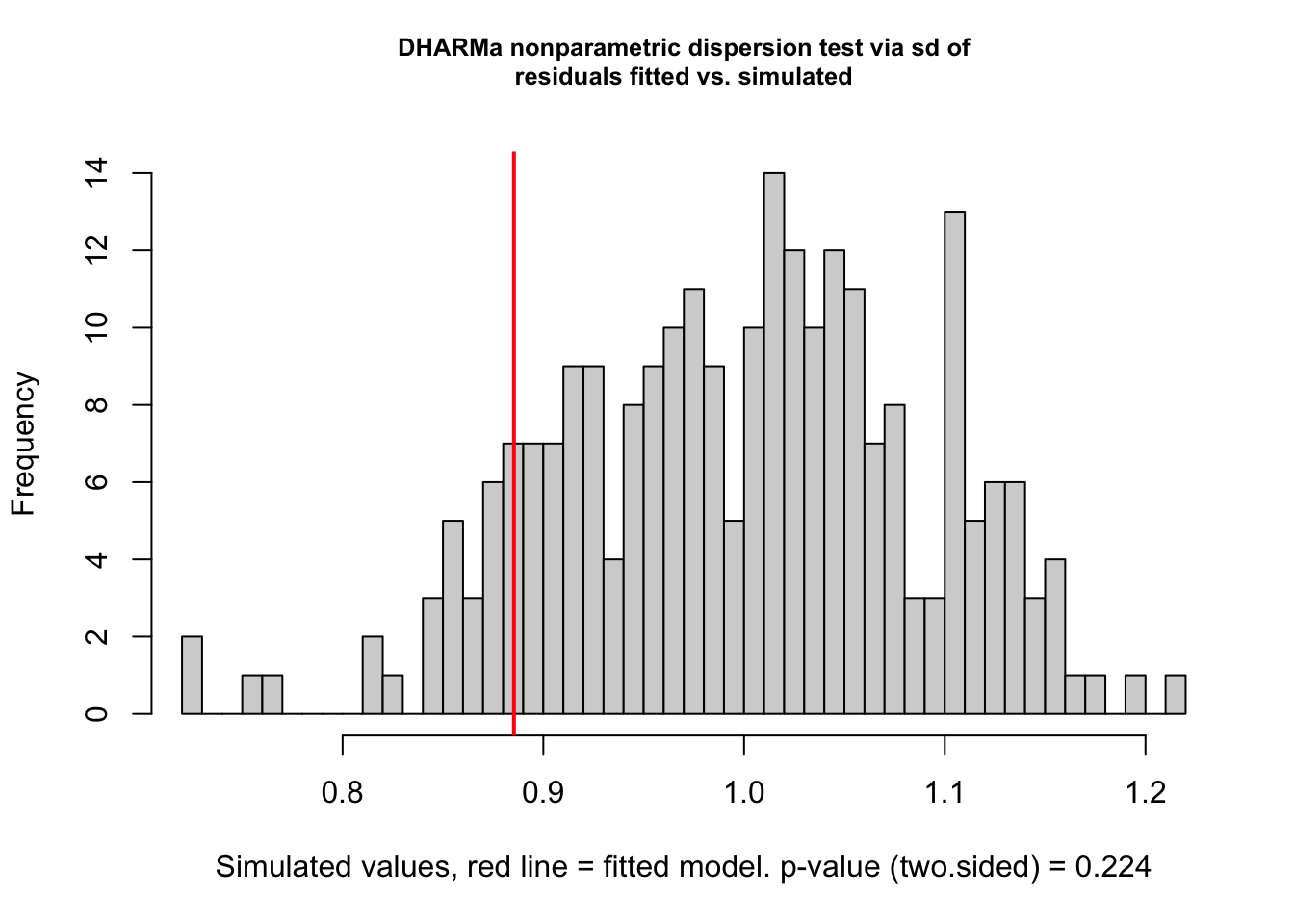
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
DHARMa nonparametric dispersion test via sd of residuals fitted vs.
simulated
data: simulationOutput
dispersion = 0.8871, p-value = 0.224
alternative hypothesis: two.sided
simulationOutput <- simulateResiduals(fittedModel = p1.mod1, plot = FALSE)
hist(residuals(simulationOutput))
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
hist(residuals(simulationOutput, quantileFunction = qnorm, outlierValues = c(-7,7)))
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
plot(simulationOutput)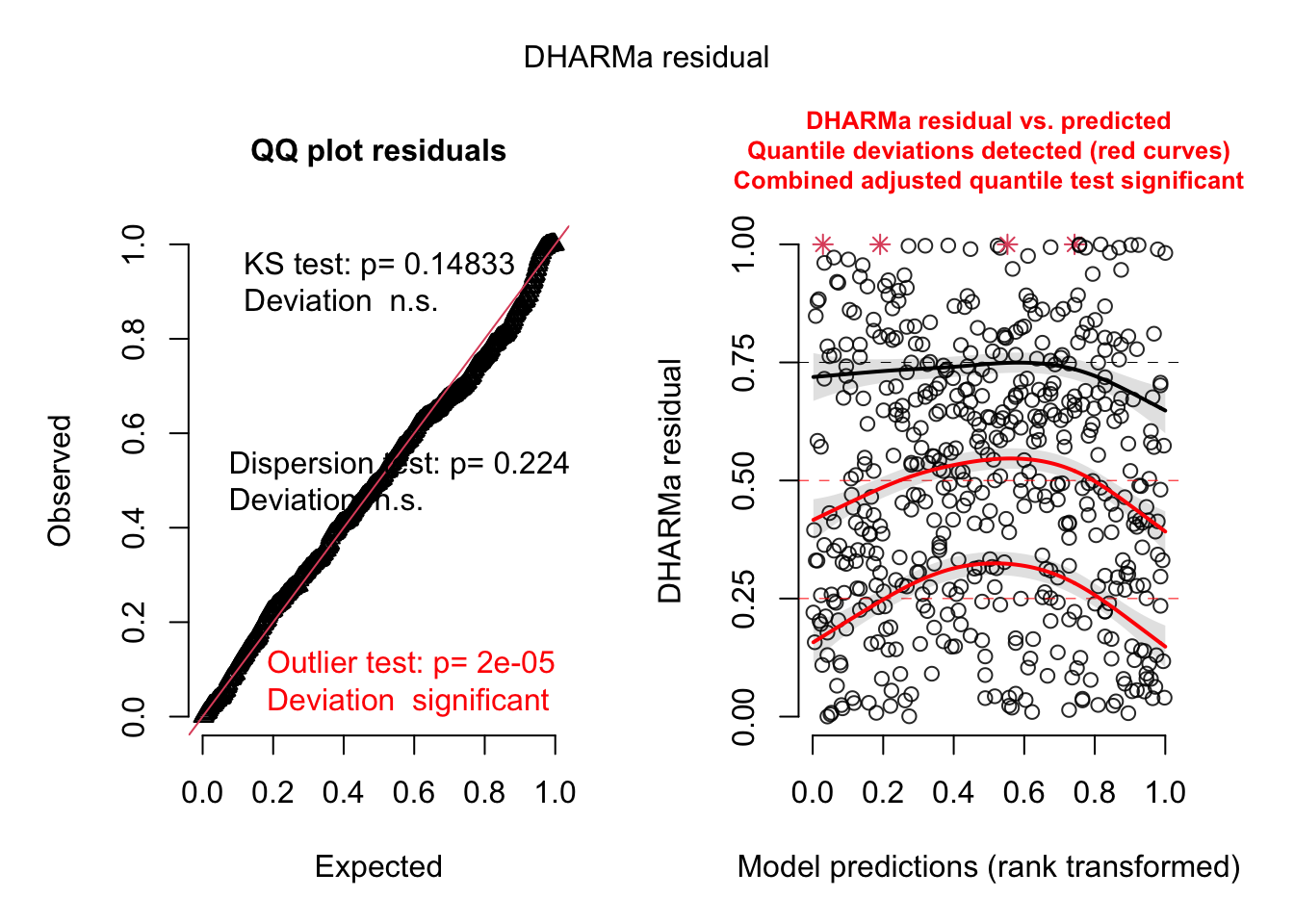
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
>>> Results
car::Anova(p1.mod1, type = "3") %>%
broom::tidy() %>%
as_tibble() %>% #write_csv("output/anova_tables/male_P1.csv") %>% # save anova table for supp. tables
kable(digits = 3,
caption = 'Analysis of Deviance Table (Type III Wald chisquare tests)') %>%
kable_styling(full_width = FALSE)| term | statistic | df | p.value |
|---|---|---|---|
| (Intercept) | 6.279 | 1 | 0.012 |
| mito | 0.574 | 2 | 0.751 |
| nuclear | 12.795 | 2 | 0.002 |
| Day | 16.861 | 1 | 0.000 |
| cop.dur | 2.209 | 1 | 0.137 |
| mito:nuclear | 2.455 | 4 | 0.653 |
# posthoc tests
bind_rows(emmeans(p1.mod1, pairwise ~ nuclear, adjust = "tukey")$contrasts %>% broom::tidy() %>%
rename(p.value = adj.p.value),
emmeans(p1.mod1, pairwise ~ Day, adjust = "tukey")$contrasts %>% broom::tidy()) %>%
mutate(p.val = ifelse(p.value < 0.001, '< 0.001', round(p.value, 3))) %>%
select(-p.value, -term) %>%
kable(digits = 3,
caption = 'Posthoc Tukey tests to compare which groups differ') %>%
kable_styling(full_width = FALSE) %>%
kableExtra::group_rows("Nuclear", 1, 3) %>%
kableExtra::group_rows("Day", 4, 4)| contrast | null.value | estimate | std.error | df | statistic | p.val |
|---|---|---|---|---|---|---|
| Nuclear | ||||||
| A - B | 0 | 0.954 | 0.272 | Inf | 3.510 | 0.001 |
| A - C | 0 | 0.291 | 0.260 | Inf | 1.118 | 0.503 |
| B - C | 0 | -0.663 | 0.269 | Inf | -2.466 | 0.036 |
| Day | ||||||
| Day1 - Day2 | 0 | 0.886 | 0.216 | Inf | 4.106 | < 0.001 |
p1plot_raw <- emmeans(p1.mod1, ~ mito * nuclear, type = 'response') %>% as_tibble() %>%
ggplot(aes(x = nuclear, y = prob, fill = mito)) +
geom_jitter(data = defence,
aes(y = P1, colour = mito),
position = position_jitterdodge(dodge.width = .5, jitter.width = .1),
alpha = .25) +
geom_jitter(data = defence %>%
group_by(LINE) %>%
summarise(mn = mean(P1)) %>%
separate(LINE, into = c("mito", "nuclear", NA), sep = "(?<=.)", remove = FALSE),
aes(y = mn, colour = mito),
position = position_jitterdodge(dodge.width = .5, jitter.width = .1),
alpha = .85, size = 2) +
geom_errorbar(aes(ymin = asymp.LCL, ymax = asymp.UCL),
width = .25, position = position_dodge(width = .5)) +
geom_point(size = 3, pch = 21, position = position_dodge(width = .5)) +
labs(y = expression(P[1]*" (EMM ± 95% CIs)")) +
scale_colour_manual(values = met3) +
scale_fill_manual(values = met3) +
theme_bw() +
theme() +
geom_text(data = defence %>%
group_by(mito, nuclear) %>% count(), aes(y = -0.04, label = n),
size = 2, position = position_dodge(width = .5)) +
#ggsave('figures/p1plot_raw.pdf', height = 4, width = 5, dpi = 600, useDingbats = FALSE) +
NULL
p1plot_raw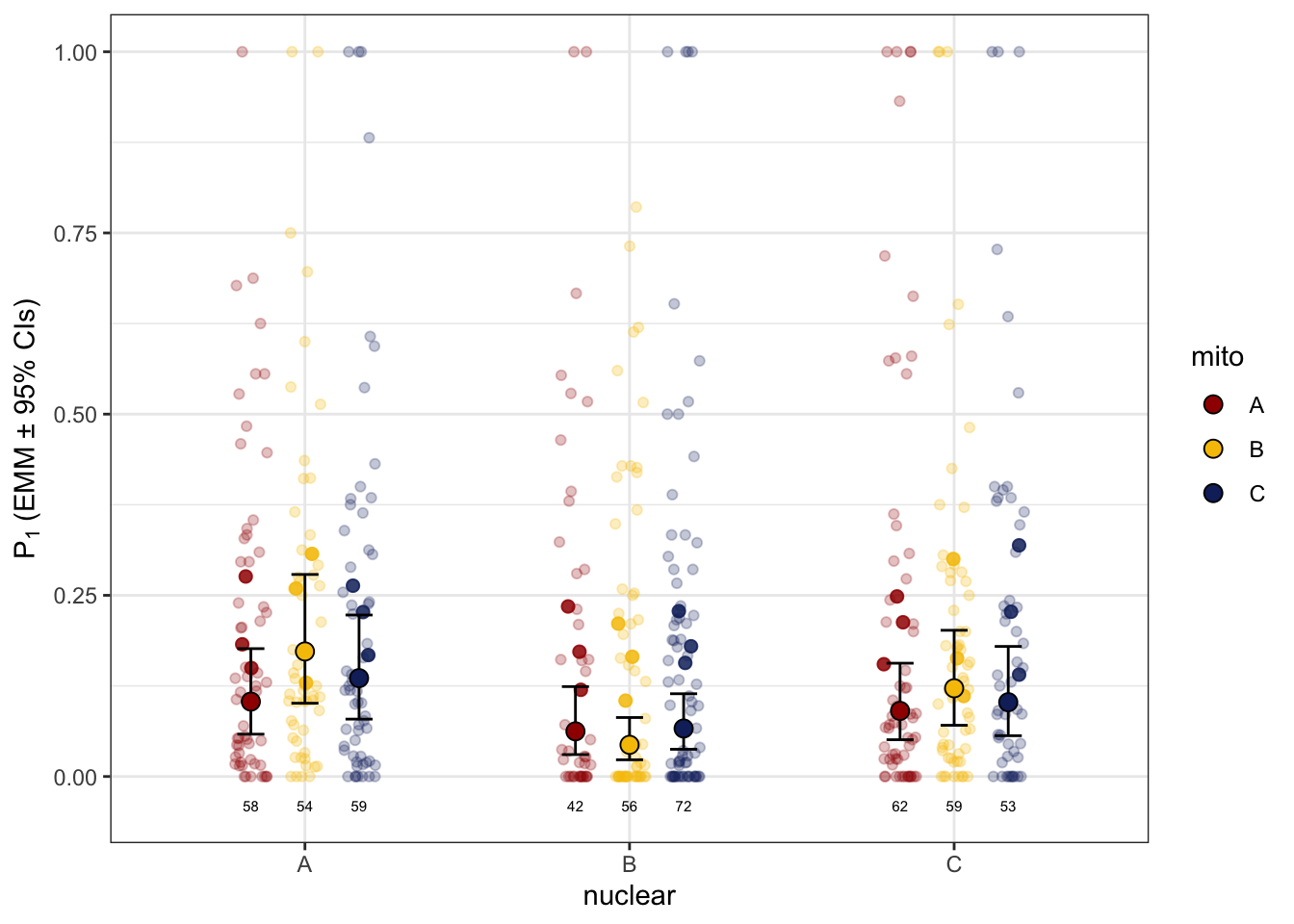
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
>>> Matched vs. mismatched
p1.coevo <- glmer(cbind(RED, BW) ~ coevolved + Day + cop.dur + (1 + cop.dur|LINE) + (1|ID) + (1|Block),
data = defence, family = binomial,
control= glmerControl(optimizer="bobyqa", optCtrl=list(maxfun=50000))
)
car::Anova(p1.coevo, type = "3")Analysis of Deviance Table (Type III Wald chisquare tests)
Response: cbind(RED, BW)
Chisq Df Pr(>Chisq)
(Intercept) 7.2827 1 0.0069623 **
coevolved 1.8058 1 0.1790107
Day 13.4235 1 0.0002485 ***
cop.dur 1.8764 1 0.1707460
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
>>> Mito-type analysis
p1.snp <- glmer(cbind(RED, BW) ~ mito_snp * nuclear + Day + cop.dur + (1 + cop.dur|LINE) + (1|ID) + (1|Block),
data = defence, family = binomial,
control= glmerControl(optimizer="bobyqa", optCtrl=list(maxfun=1e6)))
car::Anova(p1.snp, type = "3")Analysis of Deviance Table (Type III Wald chisquare tests)
Response: cbind(RED, BW)
Chisq Df Pr(>Chisq)
(Intercept) 8.0205 1 0.004625 **
mito_snp 4.9541 8 0.762472
nuclear 0.0089 2 0.995559
Day 15.2078 1 9.631e-05 ***
cop.dur 2.0267 1 0.154558
mito_snp:nuclear 8.6698 7 0.277241
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
>> Sperm offence (P2)
>>> Check covariates
>>>> Day and block effect
offence %>%
ggplot(aes(x = nuclear, y = P2, colour = mito)) +
geom_jitter(position = position_jitterdodge(dodge.width = .65, jitter.width = .15),
alpha = .5) +
labs(y = 'Sperm viability (EMM ± 95% CIs)') +
scale_colour_manual(values = met3) +
scale_fill_manual(values = met3) +
theme_bw() +
theme() +
stat_summary(position = position_dodge(width = 0.65)) +
facet_grid(Day ~ Block) +
NULL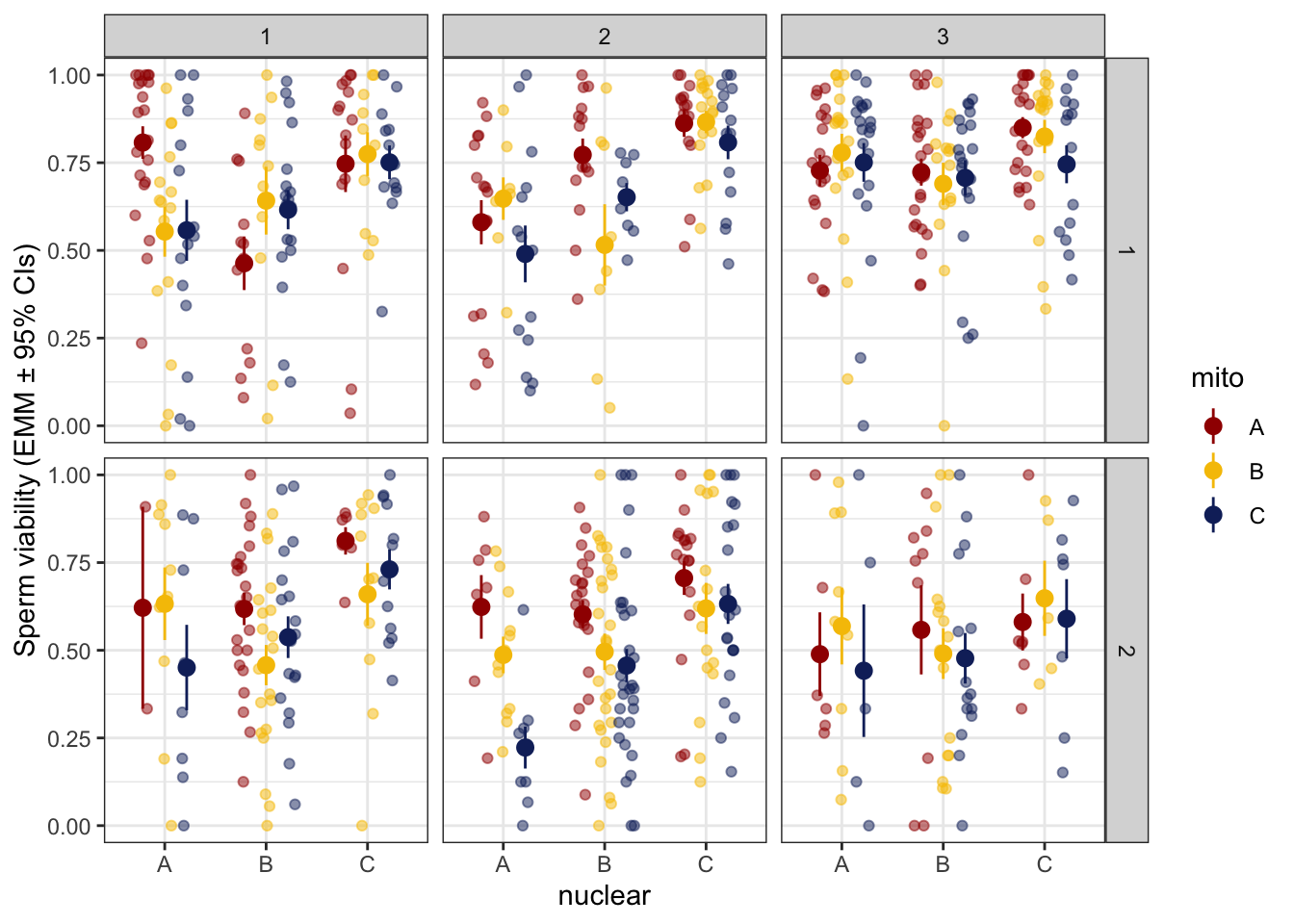
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
>>>> Copulation duration
offence %>%
ggplot(aes(x = cop.dur, y = P2, colour = nuclear)) +
geom_smooth(method = "lm", se = FALSE) +
scale_colour_manual(values = met3) +
scale_fill_manual(values = met3) +
theme_bw() +
theme() +
facet_grid(Day ~ Block) +
NULL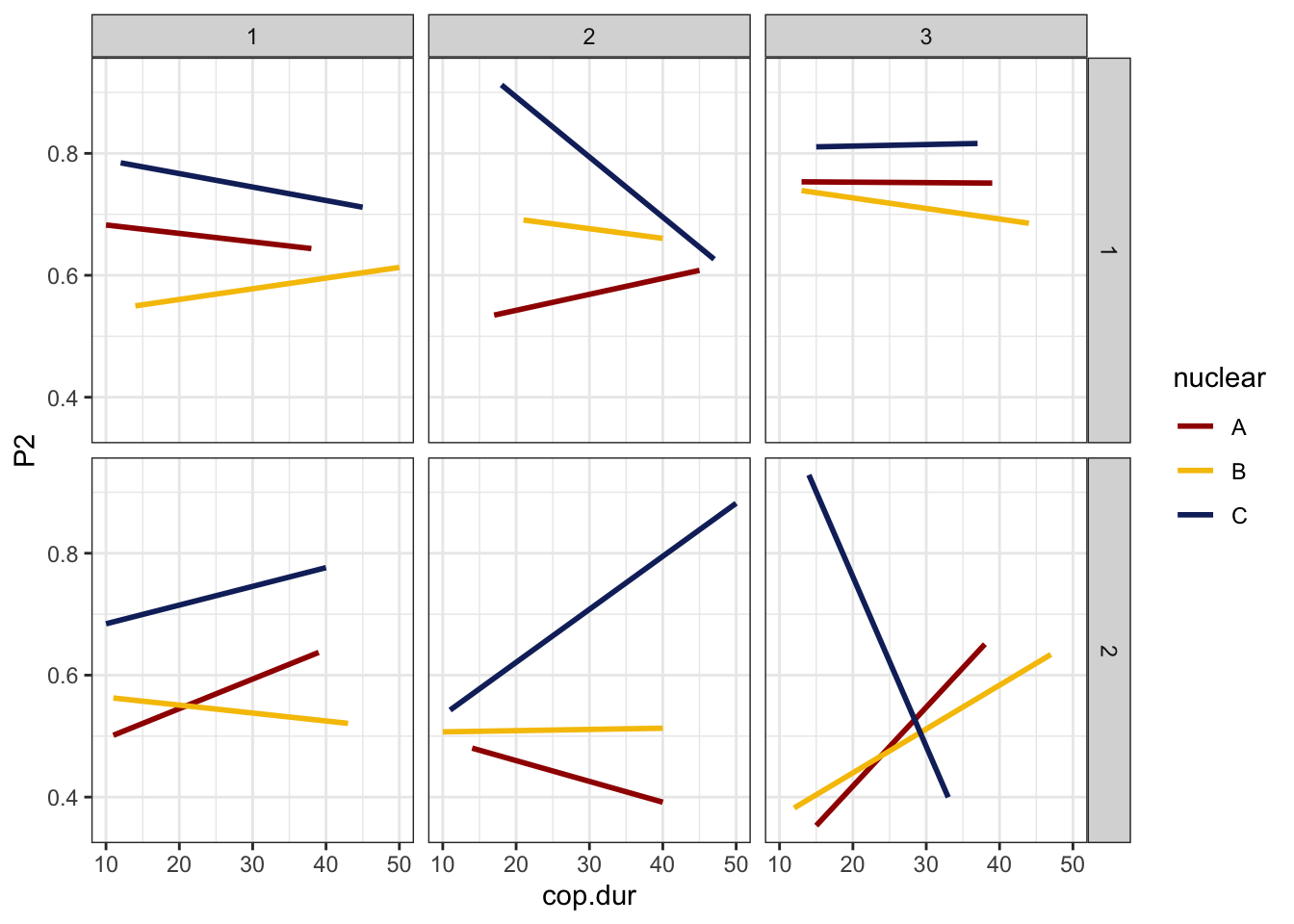
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
>>>
p2.mod1 <- glmer(cbind(RED, BW) ~ mito * nuclear + Day + cop.dur + (1 + cop.dur|LINE) + (1|ID) + (1|Block),
data = offence, family = binomial)>>>> Check diagnostics
performance::check_model(p2.mod1)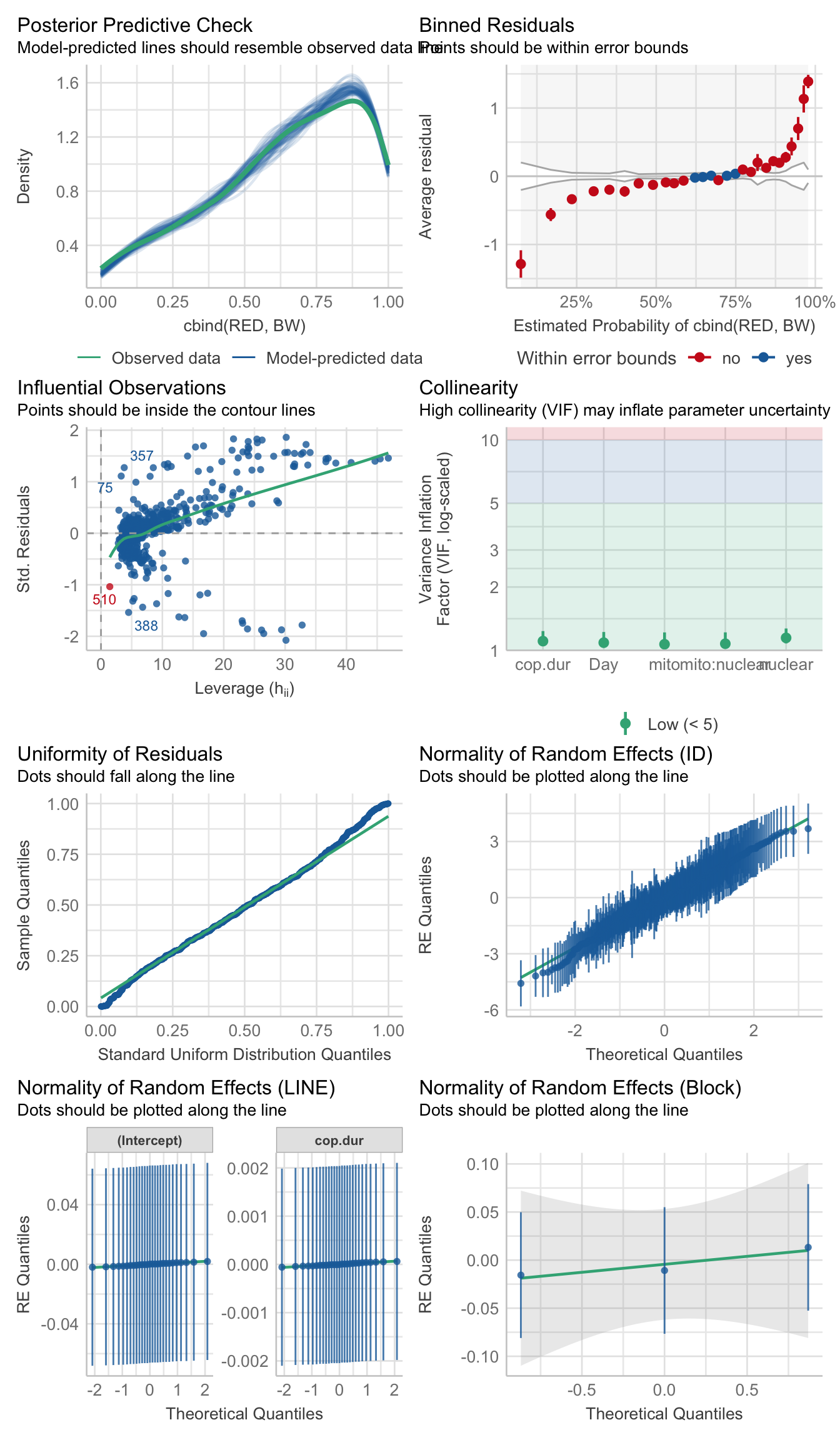
performance::check_overdispersion(p2.mod1)# Overdispersion test
dispersion ratio = 1.085
p-value = < 0.001
testDispersion(p2.mod1)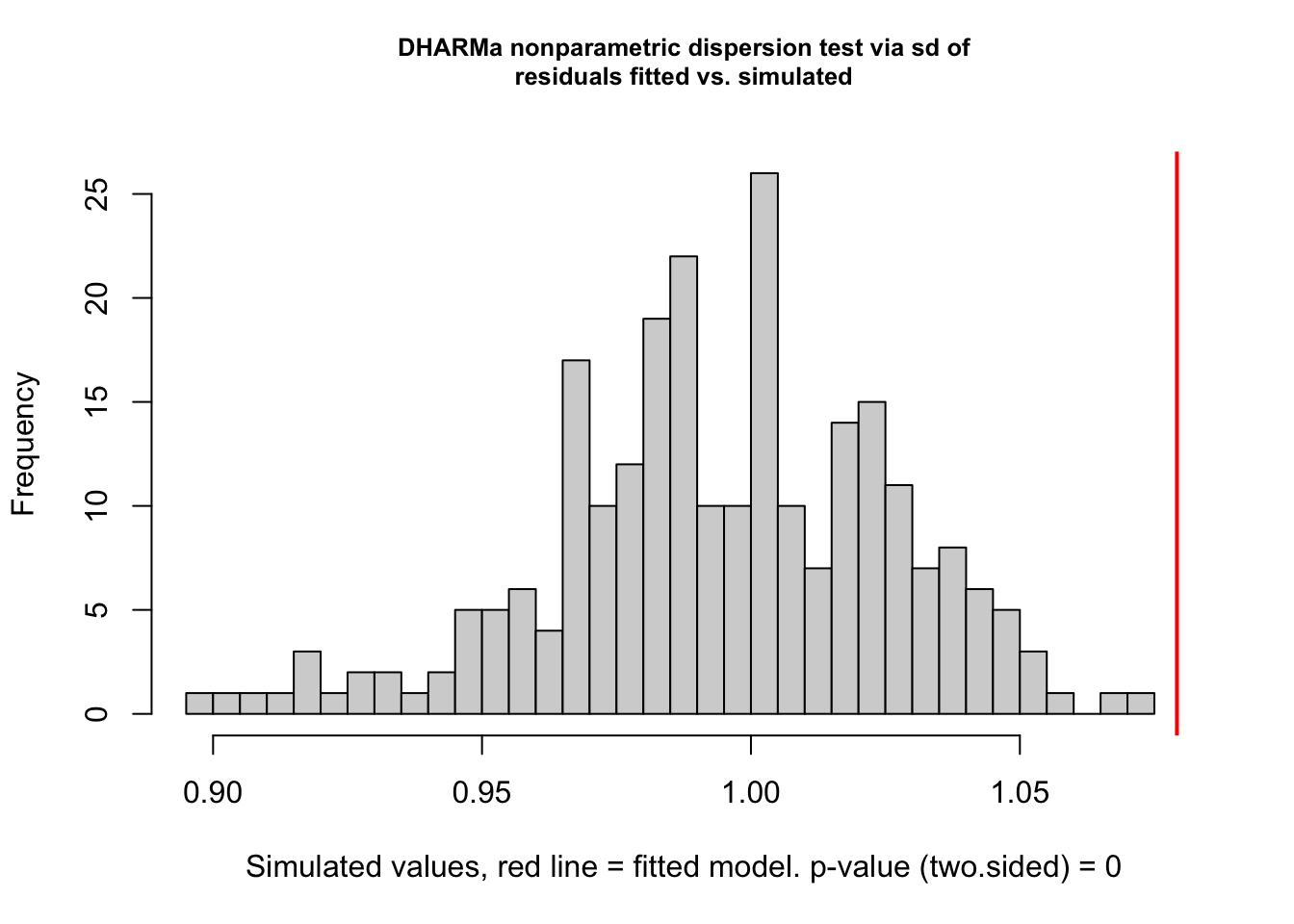
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
DHARMa nonparametric dispersion test via sd of residuals fitted vs.
simulated
data: simulationOutput
dispersion = 1.0853, p-value < 2.2e-16
alternative hypothesis: two.sided
simulationOutput <- simulateResiduals(fittedModel = p2.mod1, plot = FALSE)
hist(residuals(simulationOutput))
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
hist(residuals(simulationOutput, quantileFunction = qnorm, outlierValues = c(-7,7)))
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
plot(simulationOutput)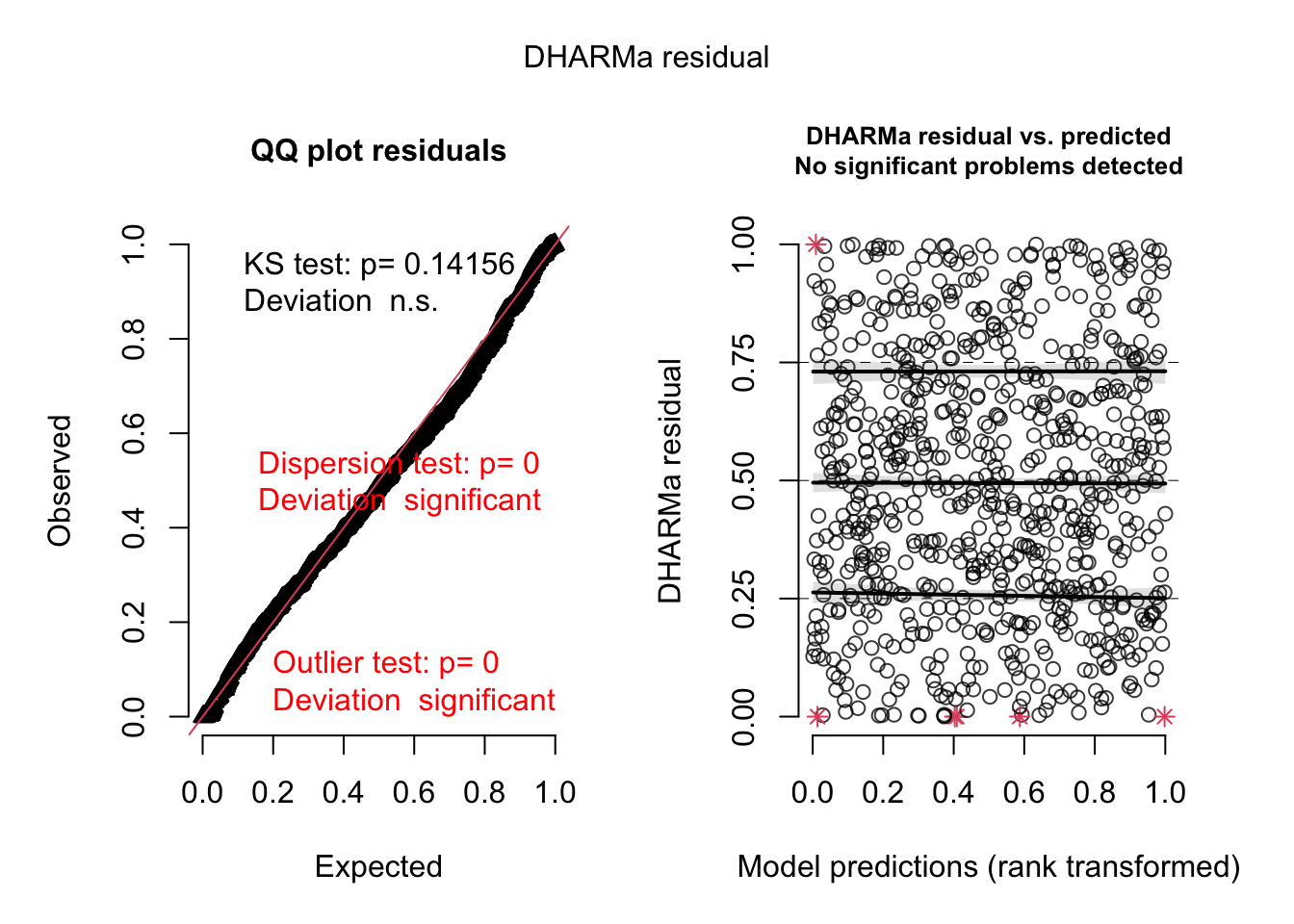
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
car::Anova(p2.mod1, type = "3") %>%
broom::tidy() %>%
as_tibble() %>% #write_csv("output/anova_tables/male_P2.csv") %>% # save anova table for supp. tables
kable(digits = 3,
caption = 'Analysis of Deviance Table (Type III Wald chisquare tests)') %>%
kable_styling(full_width = FALSE)| term | statistic | df | p.value |
|---|---|---|---|
| (Intercept) | 12.529 | 1 | 0.000 |
| mito | 10.657 | 2 | 0.005 |
| nuclear | 54.549 | 2 | 0.000 |
| Day | 49.924 | 1 | 0.000 |
| cop.dur | 0.000 | 1 | 0.988 |
| mito:nuclear | 3.681 | 4 | 0.451 |
# posthoc tests
bind_rows(emmeans(p2.mod1, pairwise ~ mito, adjust = "tukey")$contrasts %>% broom::tidy() %>%
rename(p.value = adj.p.value),
emmeans(p2.mod1, pairwise ~ nuclear, adjust = "tukey")$contrasts %>% broom::tidy() %>%
rename(p.value = adj.p.value),
emmeans(p2.mod1, pairwise ~ Day, adjust = "tukey")$contrasts %>% broom::tidy()) %>%
mutate(p.val = ifelse(p.value < 0.001, '< 0.001', round(p.value, 3))) %>%
select(-p.value, -term) %>%
kable(digits = 3,
caption = 'Posthoc Tukey tests to compare which groups differ') %>%
kable_styling(full_width = FALSE) %>%
kableExtra::group_rows("Mitochondria", 1, 3) %>%
kableExtra::group_rows("Nuclear", 4, 6) %>%
kableExtra::group_rows("Day", 7, 7)| contrast | null.value | estimate | std.error | df | statistic | p.val |
|---|---|---|---|---|---|---|
| Mitochondria | ||||||
| A - B | 0 | 0.250 | 0.141 | Inf | 1.777 | 0.177 |
| A - C | 0 | 0.455 | 0.140 | Inf | 3.260 | 0.003 |
| B - C | 0 | 0.205 | 0.140 | Inf | 1.458 | 0.311 |
| Nuclear | ||||||
| A - B | 0 | 0.027 | 0.143 | Inf | 0.191 | 0.98 |
| A - C | 0 | -0.921 | 0.149 | Inf | -6.157 | < 0.001 |
| B - C | 0 | -0.948 | 0.142 | Inf | -6.689 | < 0.001 |
| Day | ||||||
| Day1 - Day2 | 0 | 0.875 | 0.124 | Inf | 7.066 | < 0.001 |
p2plot_raw <- emmeans(p2.mod1, ~ mito * nuclear, type = 'response') %>% as_tibble() %>%
ggplot(aes(x = nuclear, y = prob, fill = mito)) +
geom_jitter(data = offence,
aes(y = P2, colour = mito),
position = position_jitterdodge(dodge.width = .5, jitter.width = .1),
alpha = .25) +
geom_jitter(data = offence %>%
group_by(LINE) %>%
summarise(mn = mean(P2)) %>%
separate(LINE, into = c("mito", "nuclear", NA), sep = "(?<=.)", remove = FALSE),
aes(y = mn, colour = mito),
position = position_jitterdodge(dodge.width = .5, jitter.width = .1),
alpha = .85, size = 2) +
geom_errorbar(aes(ymin = asymp.LCL, ymax = asymp.UCL),
width = .25, position = position_dodge(width = .5)) +
geom_point(size = 3, pch = 21, position = position_dodge(width = .5)) +
labs(y = expression(P[2]*" (EMM ± 95% CIs)")) +
scale_colour_manual(values = met3) +
scale_fill_manual(values = met3) +
theme_bw() +
theme() +
geom_text(data = defence %>%
group_by(mito, nuclear) %>% count(), aes(y = -0.04, label = n),
size = 2, position = position_dodge(width = .5)) +
#ggsave('figures/p1plot_raw.pdf', height = 4, width = 5, dpi = 600, useDingbats = FALSE) +
NULL
p2plot_raw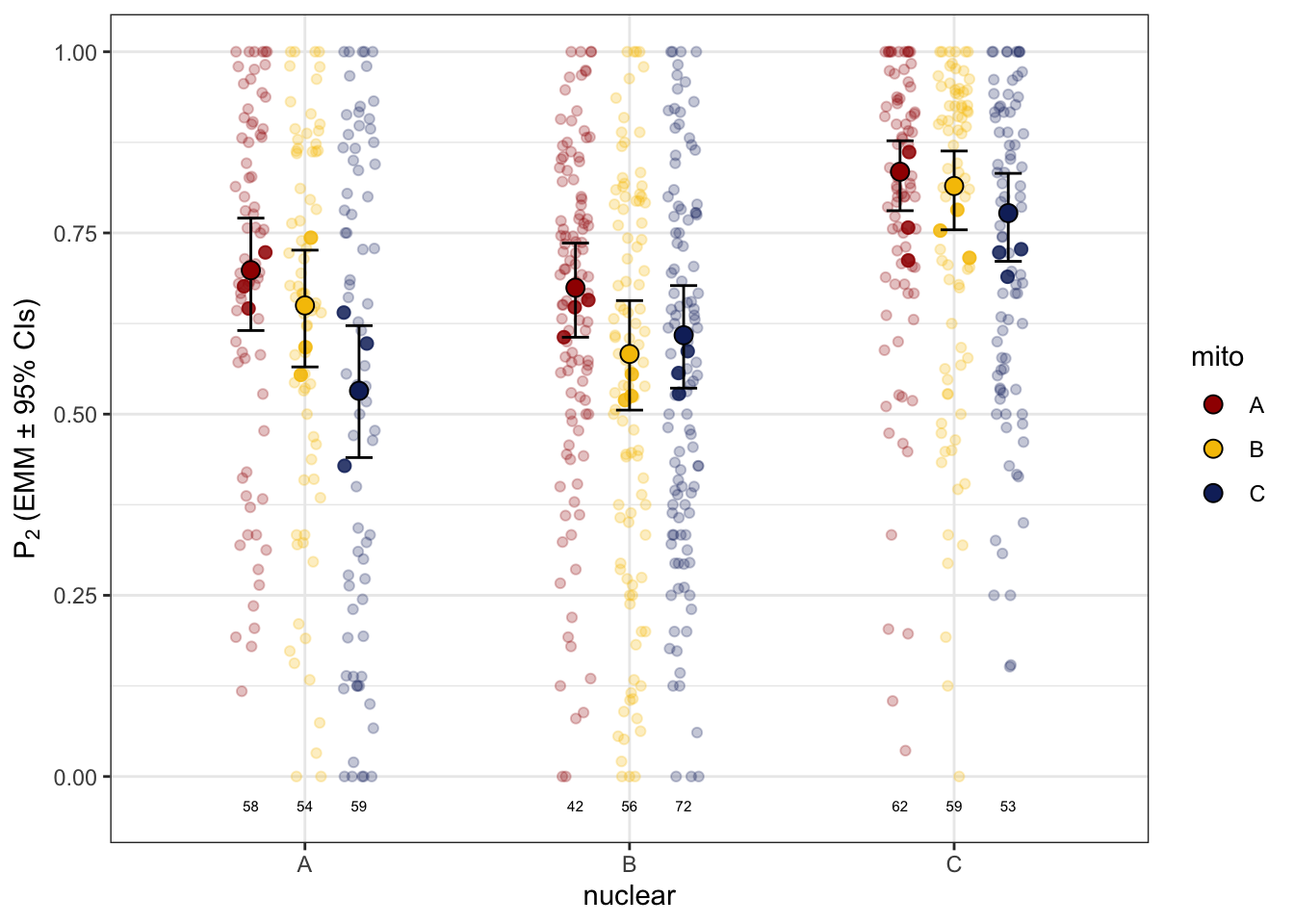
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
>>> Matched vs. mismatched
p2.coevo <- glmer(cbind(RED, BW) ~ coevolved + Day + cop.dur + (1|LINE) + (1|ID) + (1|Block),
data = offence, family = binomial,
control= glmerControl(optimizer="bobyqa", optCtrl=list(maxfun=50000)))
car::Anova(p2.coevo, type = "3")Analysis of Deviance Table (Type III Wald chisquare tests)
Response: cbind(RED, BW)
Chisq Df Pr(>Chisq)
(Intercept) 13.5083 1 0.0002375 ***
coevolved 0.0543 1 0.8157506
Day 51.4143 1 7.479e-13 ***
cop.dur 0.2653 1 0.6064804
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
>>> Mito-type analysis
p2.snp <- glmer(cbind(RED, BW) ~ mito_snp * nuclear + Day + cop.dur + (1 + cop.dur|LINE) + (1|ID) + (1|Block),
data = offence, family = binomial)
car::Anova(p2.snp, type = "3")Analysis of Deviance Table (Type III Wald chisquare tests)
Response: cbind(RED, BW)
Chisq Df Pr(>Chisq)
(Intercept) 3.0321 1 0.08163 .
mito_snp 10.0480 8 0.26167
nuclear 8.1189 2 0.01726 *
Day 44.6794 1 2.321e-11 ***
cop.dur 0.0209 1 0.88510
mito_snp:nuclear 3.8621 7 0.79551
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Combined plot
comb_spermcomp <- bind_rows(
emmeans(p1.mod1, ~ mito * nuclear, type = 'response') %>% as_tibble() %>% mutate(trt = "P1"),
emmeans(p2.mod1, ~ mito * nuclear, type = 'response') %>% as_tibble() %>% mutate(trt = "P2")) %>%
ggplot(aes(x = nuclear, y = prob, fill = mito)) +
geom_line(aes(group = mito, colour = mito), position = position_dodge(width = .5)) +
geom_point(size = 3, pch = 21, position = position_dodge(width = .5)) +
labs(y = 'Proportion sired (EMM)') +
scale_colour_manual(values = met3) +
scale_fill_manual(values = met3) +
facet_wrap(~trt) +
theme_bw() +
theme() +
NULL
comb_spermcomp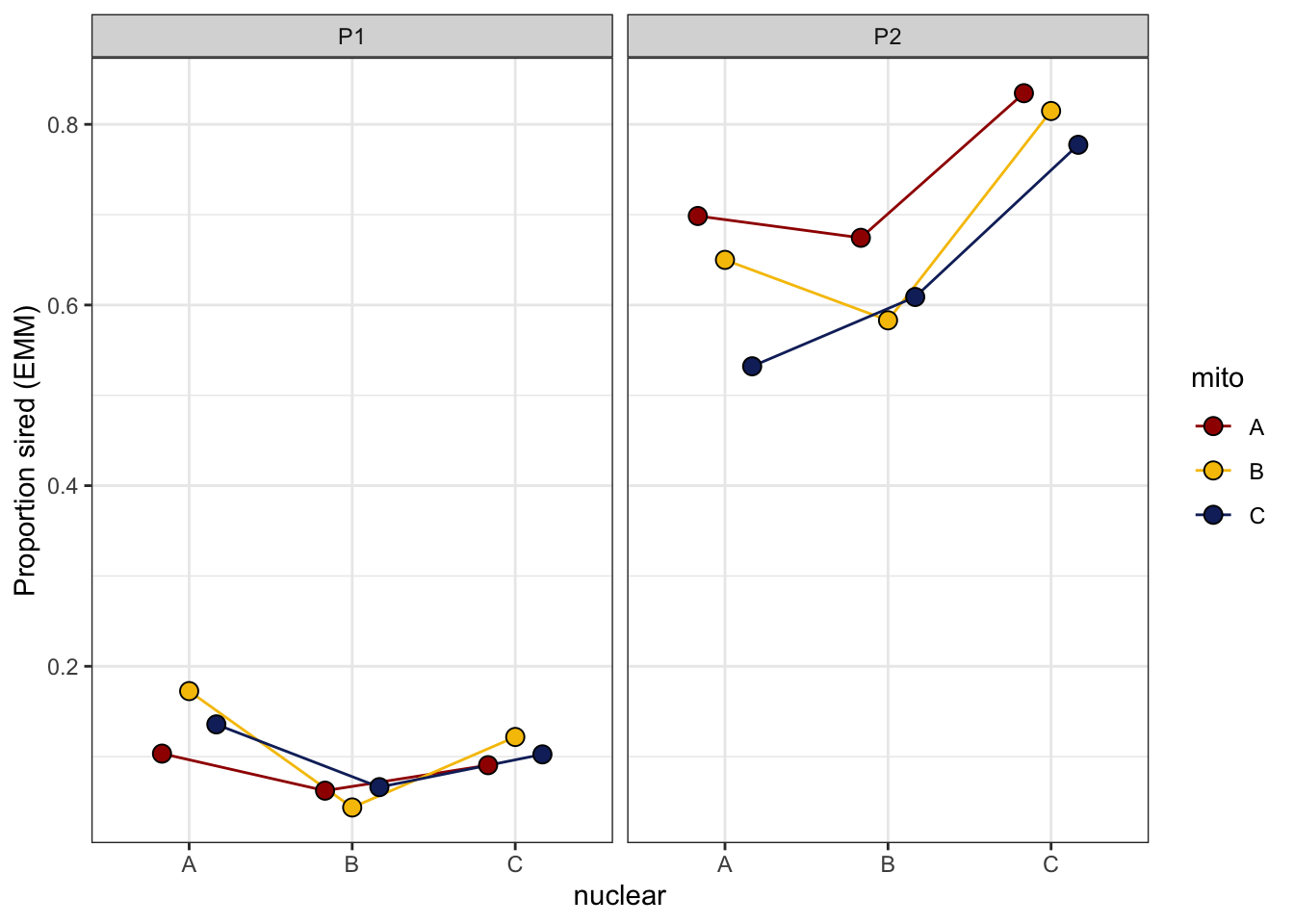
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
>> Correlation of P1 vs P2
Finally, we tested whether there was a correlation between P1 and P2 using the model estimated marginal means for each genotype. We can visualise this either as the ranked means (1-9) or as a scatterplot.
pdat <- bind_rows(emmeans(p1.mod1, ~ mito * nuclear) %>% as_tibble() %>% mutate(met = "P1"),
emmeans(p2.mod1, ~ mito * nuclear) %>% as_tibble() %>% mutate(met = "P2")) %>%
mutate(mt = rep(1:9, 2))>>> Ranked means
pdat %>%
group_by(met) %>%
mutate(rank = dense_rank(desc(emmean))) %>%
ggplot(aes(x = met, y = rank)) +
geom_point(size = 5, aes(shape = mito, colour = nuclear)) +
geom_line(aes(group = mt)) +
scale_colour_manual(values = met3) +
scale_fill_manual(values = met3) +
scale_y_continuous(breaks = 1:9) +
theme_bw()
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
>>> Scatterplot
pdat.wide <- pdat %>% select(mito, nuclear, emmean, mt, met) %>%
pivot_wider(names_from = met, values_from = emmean)
pdat.wide %>%
ggplot(aes(x = P1, y = P2)) +
geom_point(size = 5, aes(shape = mito, colour = nuclear)) +
ggpubr::stat_cor(method = "s") +
#geom_smooth(method = "lm", lty = 2, se = FALSE) +
labs(x = "logit(P1)", y = "logit(P2)") +
scale_colour_manual(values = met3) +
scale_fill_manual(values = met3) +
theme_bw() +
theme() +
#ggsave('figures/sperm_comp_cor.pdf', height = 4, width = 4.8, dpi = 600, useDingbats = FALSE) +
NULL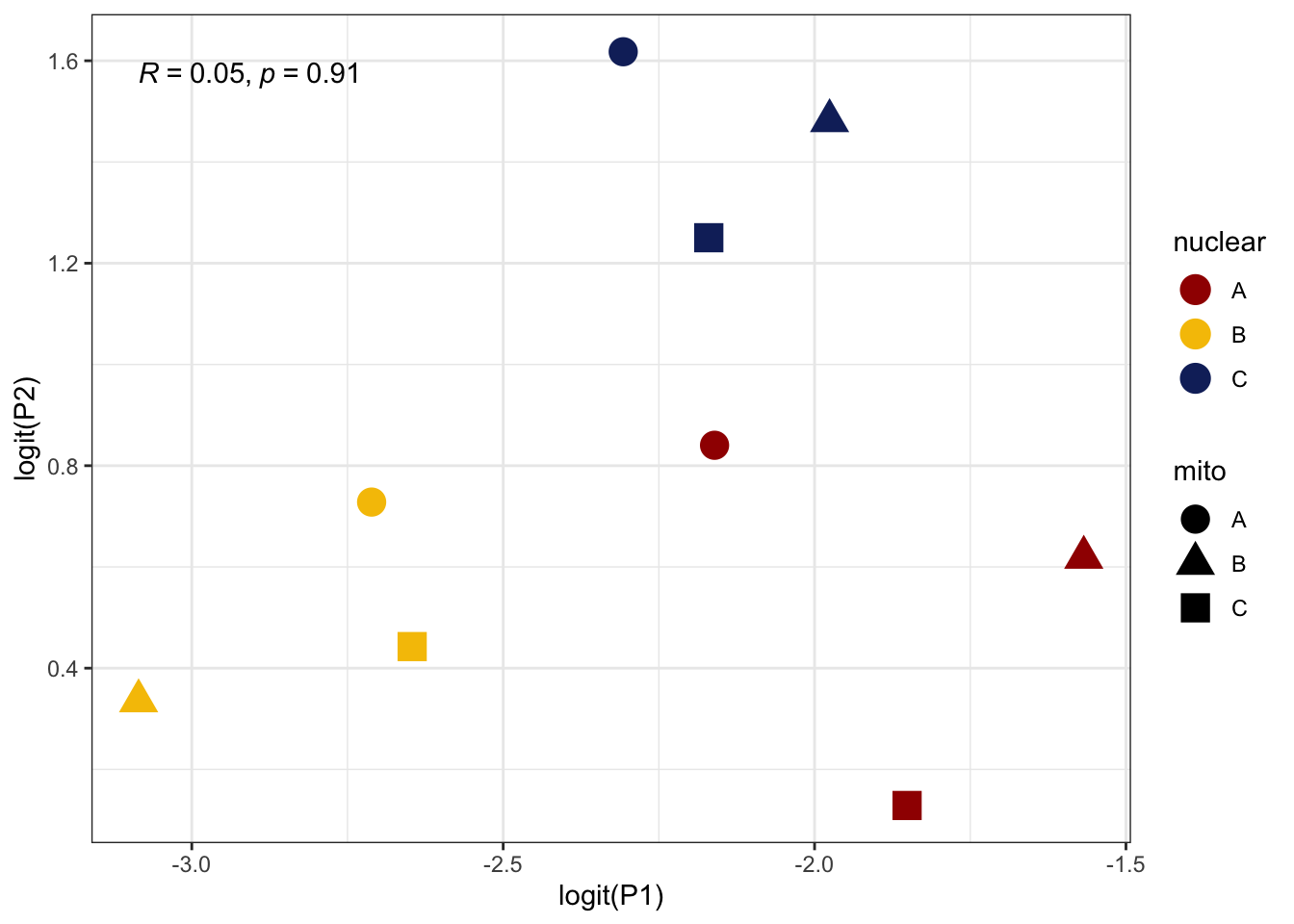
| Version | Author | Date |
|---|---|---|
| a7bae31 | MartinGarlovsky | 2024-10-09 |
cor.test(pdat.wide$P1, pdat.wide$P2, method = "s")
Spearman's rank correlation rho
data: pdat.wide$P1 and pdat.wide$P2
S = 114, p-value = 0.9116
alternative hypothesis: true rho is not equal to 0
sample estimates:
rho
0.05
sessionInfo()R version 4.4.0 (2024-04-24) Platform: aarch64-apple-darwin20 Running under: macOS Sonoma 14.6.1 Matrix products: default BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0 locale: [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8 time zone: Europe/London tzcode source: internal attached base packages: [1] stats graphics grDevices utils datasets methods base other attached packages: [1] conflicted_1.2.0 showtext_0.9-7 showtextdb_3.0 sysfonts_0.8.9 [5] knitrhooks_0.0.4 knitr_1.48 kableExtra_1.4.0 emmeans_1.10.5 [9] DHARMa_0.4.7 lme4_1.1-35.5 Matrix_1.7-1 lubridate_1.9.3 [13] forcats_1.0.0 stringr_1.5.1 dplyr_1.1.4 purrr_1.0.2 [17] readr_2.1.5 tidyr_1.3.1 tibble_3.2.1 ggplot2_3.5.1 [21] tidyverse_2.0.0 workflowr_1.7.1 loaded via a namespace (and not attached): [1] Rdpack_2.6.1 rlang_1.1.4 magrittr_2.0.3 [4] git2r_0.35.0 compiler_4.4.0 getPass_0.2-4 [7] mgcv_1.9-1 systemfonts_1.1.0 callr_3.7.6 [10] vctrs_0.6.5 pkgconfig_2.0.3 fastmap_1.2.0 [13] backports_1.5.0 MetBrewer_0.2.0 labeling_0.4.3 [16] utf8_1.2.4 promises_1.3.0 rmarkdown_2.28 [19] tzdb_0.4.0 ps_1.8.1 nloptr_2.1.1 [22] xfun_0.48 cachem_1.1.0 jsonlite_1.8.9 [25] highr_0.11 later_1.3.2 broom_1.0.7 [28] parallel_4.4.0 R6_2.5.1 gap.datasets_0.0.6 [31] qgam_1.3.4 bslib_0.8.0 stringi_1.8.4 [34] car_3.1-3 boot_1.3-31 jquerylib_0.1.4 [37] numDeriv_2016.8-1.1 estimability_1.5.1 iterators_1.0.14 [40] Rcpp_1.0.13 httpuv_1.6.15 splines_4.4.0 [43] timechange_0.3.0 tidyselect_1.2.1 abind_1.4-8 [46] rstudioapi_0.17.1 yaml_2.3.10 codetools_0.2-20 [49] doParallel_1.0.17 processx_3.8.4 plyr_1.8.9 [52] lattice_0.22-6 lmerTest_3.1-3 shiny_1.9.1 [55] bayestestR_0.15.0 withr_3.0.2 coda_0.19-4.1 [58] evaluate_1.0.1 xml2_1.3.6 ggpubr_0.6.0 [61] pillar_1.9.0 carData_3.0-5 gap_1.6 [64] whisker_0.4.1 foreach_1.5.2 insight_1.0.1 [67] generics_0.1.3 rprojroot_2.0.4 hms_1.1.3 [70] munsell_0.5.1 scales_1.3.0 minqa_1.2.8 [73] xtable_1.8-4 glue_1.8.0 tools_4.4.0 [76] see_0.9.0 ggsignif_0.6.4 fs_1.6.5 [79] mvtnorm_1.3-1 grid_4.4.0 rbibutils_2.3 [82] datawizard_1.0.0 colorspace_2.1-1 nlme_3.1-166 [85] patchwork_1.3.0 performance_0.13.0 Formula_1.2-5 [88] cli_3.6.3 fansi_1.0.6 viridisLite_0.4.2 [91] svglite_2.1.3 gtable_0.3.6 rstatix_0.7.2 [94] sass_0.4.9 digest_0.6.37 pbkrtest_0.5.3 [97] ggrepel_0.9.6 farver_2.1.2 memoise_2.0.1 [100] htmltools_0.5.8.1 lifecycle_1.0.4 httr_1.4.7 [103] mime_0.12 MASS_7.3-61